
Latest recommendations

| Id | Title | Authors | Abstract▲ | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
18 Nov 2020
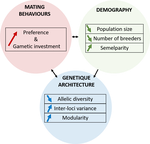
A demogenetic agent based model for the evolution of traits and genome architecture under sexual selectionSexual selection goes dynamicRecommended by Michael D Greenfield based on reviews by Frédéric Guillaume and 1 anonymous reviewer150 years after Darwin published ‘Descent of man and selection in relation to sex’ (Darwin, 1871), the evolutionary mechanism that he laid out in his treatise continues to fascinate us. Sexual selection is responsible for some of the most spectacular traits among animals, and plants, and it appeals to our interest in all things reproductive and sexual (Bell, 1982). In addition, sexual selection poses some of the more intractable problems in evolutionary biology: Its realm encompasses traits that are subject to markedly different selection pressures, particularly when distinct, yet associated, traits tend to be associated with males, e.g. courtship signals, and with females, e.g. preferences (cf. Ah-King & Ahnesjo, 2013). While separate, such traits cannot evolve independently of each other (Arnqvist & Rowe, 2005), and complex feedback loops and correlations between them are predicted (Greenfield et al., 2014). Traditionally, sexual selection has been modelled under simplifying assumptions, and quantitative genetic approaches that avoided evolutionary dynamics have prevailed. New computing methods may be able to free the field from these constraints, and a trio of theoreticians (Chevalier, De Coligny & Labonne 2020) describe here a novel application of a ‘demo-genetic agent (or individual) based model’, a mouthful hereafter termed DG-ABM, for arriving at a holistic picture of the sexual selection trajectory. The application is built on the premise that traits, e.g. courtship, preference, gamete investment, competitiveness for mates, can influence the genetic architecture, e.g. correlations, of those traits. In turn, the genetic architecture can influence the expression and evolvability of the traits. Much of this influence occurs via demographic features, i.e. social environment, generated by behavioral interactions during sexual advertisement, courtship, mate guarding, parental care, post-mating dispersal, etc. References Ah-King, M. and Ahnesjo, I. 2013. The ‘sex role’ concept: An overview and evaluation Evolutionary Biology, 40, 461-470. doi: https://doi.org/10.1007/s11692-013-9226-7 | A demogenetic agent based model for the evolution of traits and genome architecture under sexual selection | Louise Chevalier, François de Coligny, Jacques Labonne | <p>Sexual selection has long been known to favor the evolution of mating behaviors such as mate preference and competitiveness, and to affect their genetic architecture, for instance by favoring genetic correlation between some traits. Reciprocall... | | Adaptation, Behavior & Social Evolution, Evolutionary Dynamics, Evolutionary Theory, Life History, Population Genetics / Genomics, Sexual Selection | Michael D Greenfield | 2020-04-02 14:44:25 | ||
04 Mar 2021
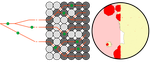
Simulation of bacterial populations with SLiMSimulating bacterial evolution forward-in-timeRecommended by Frederic Bertels based on reviews by 3 anonymous reviewersJean Cury and colleagues (2021) have developed a protocol to simulate bacterial evolution in SLiM. In contrast to existing methods that depend on the coalescent, SLiM simulates evolution forward in time. SLiM has, up to now, mostly been used to simulate the evolution of eukaryotes (Haller and Messer 2019), but has been adapted here to simulate evolution in bacteria. Forward-in-time simulations are usually computationally very costly. To circumvent this issue, bacterial population sizes are scaled down. One would now expect results to become inaccurate, however, Cury et al. show that scaled-down forwards simulations provide very accurate results (similar to those provided by coalescent simulators) that are consistent with theoretical expectations. Simulations were analyzed and compared to existing methods in simple and slightly more complex scenarios where recombination affects evolution. In all scenarios, simulation results from coalescent methods (fastSimBac (De Maio and Wilson 2017), ms (Hudson 2002)) and scaled-down forwards simulations were very similar, which is very good news indeed. A biologist not aware of the complexities of forwards, backwards simulations and the coalescent, might now naïvely ask why another simulation method is needed if existing methods perform just as well. To address this question the manuscript closes with a very neat example of what exactly is possible with forwards simulations that cannot be achieved using existing methods. The situation modeled is the growth and evolution of a set of 50 bacteria that are randomly distributed on a petri dish. One side of the petri dish is covered in an antibiotic the other is antibiotic-free. Over time, the bacteria grow and acquire antibiotic resistance mutations until the entire artificial petri dish is covered with a bacterial lawn. This simulation demonstrates that it is possible to simulate extremely complex (e.g. real world) scenarios to, for example, assess whether certain phenomena are expected with our current understanding of bacterial evolution, or whether there are additional forces that need to be taken into account. Hence, forwards simulators could significantly help us to understand what current models can and cannot explain in evolutionary biology.
References Cury J, Haller BC, Achaz G, Jay F (2021) Simulation of bacterial populations with SLiM. bioRxiv, 2020.09.28.316869, version 5 peer-reviewed and recommended by Peer community in Evolutionary Biology. https://doi.org/10.1101/2020.09.28.316869 De Maio N, Wilson DJ (2017) The Bacterial Sequential Markov Coalescent. Genetics, 206, 333–343. https://doi.org/10.1534/genetics.116.198796 Haller BC, Messer PW (2019) SLiM 3: Forward Genetic Simulations Beyond the Wright–Fisher Model. Molecular Biology and Evolution, 36, 632–637. https://doi.org/10.1093/molbev/msy228 Hudson RR (2002) Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics, 18, 337–338. https://doi.org/10.1093/bioinformatics/18.2.337 | Simulation of bacterial populations with SLiM | Jean Cury, Benjamin C. Haller, Guillaume Achaz, and Flora Jay | <p>Simulation of genomic data is a key tool in population genetics, yet, to date, there is no forward-in-time simulator of bacterial populations that is both computationally efficient and adaptable to a wide range of scenarios. Here we demonstrate... | | Bioinformatics & Computational Biology, Population Genetics / Genomics | Frederic Bertels | 2020-10-02 19:03:42 | ||
25 Jun 2020

Transcriptional differences between the two host strains of Spodoptera frugiperda (Lepidoptera: Noctuidae)Speciation through selection on mitochondrial genes?Recommended by Astrid Groot based on reviews by Heiko Vogel and Sabine HaennigerWhether speciation through ecological specialization occurs has been a thriving research area ever since Mayr (1942) stated this to play a central role. In herbivorous insects, ecological specialization is most likely to happen through host plant differentiation (Funk et al. 2002). Therefore, after Dorothy Pashley had identified two host strains in the Fall armyworm (FAW), Spodoptera frugiperda, in 1988 (Pashley 1988), researchers have been trying to decipher the evolutionary history of these strains, as this seems to be a model species in which speciation is currently occurring through host plant specialization. Even though FAW is a generalist, feeding on many different host plant species (Pogue 2002) and a devastating pest in many crops, Pashley identified a so-called corn strain and a so-called rice strain in Puerto Rico. Genetically, these strains were found to differ mostly in an esterase, although later studies showed additional genetic differences and markers, mostly in the mitochondrial COI and the nuclear TPI. Recent genomic studies showed that the two strains are overall so genetically different (2% of their genome being different) that these two strains could better be called different species (Kergoat et al. 2012). So far, the most consistent differences between the strains have been their timing of mating activities at night (Schoefl et al. 2009, 2011; Haenniger et al. 2019) and hybrid incompatibilities (Dumas et al. 2015; Kost et al. 2016). Whether and to what extent host plant preference or performance contributed to the differentiation of these sympatrically occurring strains has remained unclear. References [1] Dumas, P. et al. (2015). Spodoptera frugiperda (Lepidoptera: Noctuidae) host-plant variants: two host strains or two distinct species?. Genetica, 143(3), 305-316. doi: 10.1007/s10709-015-9829-2 | Transcriptional differences between the two host strains of Spodoptera frugiperda (Lepidoptera: Noctuidae) | Marion Orsucci, Yves Moné, Philippe Audiot, Sylvie Gimenez, Sandra Nhim, Rima Naït-Saïdi, Marie Frayssinet, Guillaume Dumont, Jean-Paul Boudon, Marin Vabre, Stéphanie Rialle, Rachid Koual, Gael J. Kergoat, Rodney N. Nagoshi, Robert L. Meagher, Emm... | <p>Spodoptera frugiperda, the fall armyworm (FAW), is an important agricultural pest in the Americas and an emerging pest in sub-Saharan Africa, India, East-Asia and Australia, causing damage to major crops such as corn, sorghum and soybean. While... | | Adaptation, Evolutionary Ecology, Expression Studies, Life History, Speciation | Astrid Groot | 2018-05-09 13:04:34 | ||
11 Oct 2022
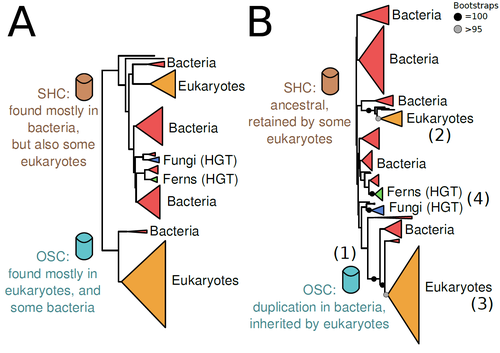
The Eukaryotic Last Common Ancestor Was Bifunctional for Hopanoid and Sterol ProductionGene family analysis suggests new evolutionary scenario for sterol and hopanoid biomarkersRecommended by Iker Irisarri based on reviews by Samuel Abalde, Denis Baurain and Jose Ramon Pardos-BlasSterols and hopanoids are sometimes used as biomarkers to infer the origin of certain groups of organisms. Traditionally, hopanoid-derived products in ancient rocks have been considered to indicate the presence of bacteria, whereas sterol derivatives have been considered to be exclusive to eukaryotes. However, a closer look at the topic reveals a rather complex distribution of either compound in both bacteria and eukaryotes. (1). The known biosynthetic pathways for sterols and hopanoids are similar but diverge at a critical step where two different enzymes are used: squalene-hopene cyclase (SHC) and oxidosqualene cyclase (OSC), the latter requiring oxygen. These two enzymes belong to the same gene family, whose complex evolutionary history is difficult to reconcile with the known species phylogeny. In this study (2), Dr. Warren R. Francis revisits the evolution of this gene family using an extended dataset with a broader taxonomic representation. In contrast to the traditional representation of the tree rooted between SHC and OSC paralogs (i.e., based on function), the author proposes that rooting the tree within bacterial SHCs and assuming a secondary origin of OSC is more parsimonious. This postulates SHC to be the ancestral function –retained in many extant bacteria and some eukaryotes– and OSC to have emerged later within bacteria –currently being mostly present in eukaryotes–. The reconstructed evolutionary history is arguably complex and can only be reconciled with the species' phylogeny by invoking many secondary losses. These losses are considered likely because many extant species acquire sterols and hopanoids by diet and lack one or both enzymes. Some cases of recent horizontal gene transfer are also proposed. In contrast to the dichotomy between bacterial SHCs and eukaryote OSCs, the new proposed scenario suggests that the eukaryote ancestor likely inherited both enzymes from bacteria and thus could be able to synthesize both sterols and hopanoids. Under this hypothesis, not only bacteria but also eukaryotes could be responsible for the hopane found in old rocks. This agrees with eukaryote fossils dating back to more than 1 billion years ago (3). Also, the observed increase of sterane levels in rocks ~600-700 million years old cannot be associated with the origin of eukaryotes, which is a much older event, but could rather reflect changes in atmospheric oxygen levels because oxygen is required for the synthesis of sterols by OSC. References 1. Santana-Molina C, Rivas-Marin E, Rojas AM, Devos DP (2020) Origin and Evolution of Polycyclic Triterpene Synthesis. Molecular Biology and Evolution, 37, 1925–1941. https://doi.org/10.1093/molbev/msaa054 2. Francis WR (2022) The Eukaryotic Last Common Ancestor Was Bifunctional for Hopanoid and Sterol Production. Preprints, 2020040186, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.20944/preprints202004.0186.v5 3. Butterfield NJ (2000) Bangiomorpha pubescens n. gen., n. sp.: implications for the evolution of sex, multicellularity, and the Mesoproterozoic/Neoproterozoic radiation of eukaryotes. Paleobiology, 26, 386–404. https://doi.org/10.1666/0094-8373(2000)026<0386:BPNGNS>2.0.CO;2 | The Eukaryotic Last Common Ancestor Was Bifunctional for Hopanoid and Sterol Production | Warren R Francis | <p>Steroid and hopanoid biomarkers can be found in ancient rocks and may give a glimpse of what life was present at that time. Sterols and hopanoids are produced by two related enzymes, though the evolutionary history of this protein family is com... | | Bioinformatics & Computational Biology, Evolutionary Ecology, Molecular Evolution, Paleontology, Phylogenetics / Phylogenomics | Iker Irisarri | 2021-01-13 16:03:29 | ||
02 May 2023
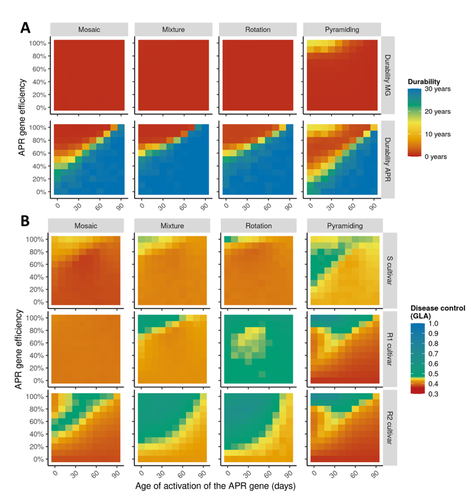
Durable resistance or efficient disease control? Adult Plant Resistance (APR) at the heart of the dilemmaPlant resistance to pathogens: just you wait?Recommended by Timothée Poisot based on reviews by Jean-Paul Soularue and 1 anonymous reviewerIn this preprint, Rimbaud et al. (2023) examine whether Adult Plant Resistance (APR), where plants delay their response to pathogens, is a viable alternative when the solution to evolve complete resistance from the seedling stage exists. At first glance, delaying resistance seems like a counter-intuitive strategy, unless it can result in a weaker selection of the pathogen, and therefore slow down its adaption to plant resistance. The approach of Rimbaud et al. is to incorporate as much of the mechanisms as possible into a model. By accounting for explicit spatio-temporal dynamics, stochasticity, and the coupling between demography and population genetics, to simulate an agricultural landscape, they reach a nuanced conclusion. Weaker and delayed activation of genes that confer APR does indeed reduce the selection pressure acting on the pathogen, at the cost of overall less effective protection. The alternative strategy of rapid or complete activation of these genes, although it results in better results in defending against the pathogen, is at risk of being overcome because it introduces a stronger selection pressure. One important feature of this work is that it accounts for agricultural practices. The landscape that is simulated can account for monoculture, mosaic cultures, mixed cultures, and rotations of crops (with different strategies for resistance). This introduces an interesting element to the conclusion: that human practices will have an impact on the selection pressures acting within the system. Perhaps the most striking result is that, for the plants, it might be more beneficial to bear the cost of a wild-type pathogen that can benefit from delayed activation of resistance, and therefore exclude the more virulent strains by simply being there first, and essentially buying the plant some time before it activates its resistance more completely. When the landscape is aggregated, even wild-type pathogens can cause severe epidemics; increasing fragmentation, because it enables connectivity between patches of plants with different strategies, allows pathogens to move across cultivars, and reduces the epidemic risk on susceptible plants. These results should encourage scaling up the perspective on APR, and indeed Rimbaud et al. adopt a landscape-scale perspective, to show that APR genes and genes conferring more complete resistance early on can have synergistic effects. This is, again, both an interesting result for evolutionary biologists, but also a useful way to prioritize different crop management strategies over large spatial scales. References Rimbaud, Loup, et al. Durable Resistance or Efficient Disease Control? Adult Plant Resistance (APR) at the Heart of the Dilemma. 2023. bioRxiv, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.08.30.505787 | Durable resistance or efficient disease control? Adult Plant Resistance (APR) at the heart of the dilemma | Loup Rimbaud, Julien Papaïx, Jean-François Rey, Benoît Moury, Luke G. Barrett, Peter H. Thrall | <p style="text-align: justify;">Adult plant resistance (APR) is an incomplete and delayed protection of plants against pathogens. At first glance, such resistance should be less efficient than classical major-effect resistance genes, which confer ... | | Adaptation, Evolutionary Applications, Evolutionary Epidemiology | Timothée Poisot | 2022-09-02 16:36:32 | ||
11 May 2023

Co-obligate symbioses have repeatedly evolved across aphids, but partner identity and nutritional contributions vary across lineagesFlexibility in Aphid Endosymbiosis: Dual Symbioses Have Evolved Anew at Least Six TimesRecommended by Olivier Tenaillon based on reviews by Alex C. C. Wilson and 1 anonymous reviewerIn this intriguing study (Manzano-Marín et al. 2022) by Alejandro Manzano-Marin and his colleagues, the association between aphids and their symbionts is investigated through meta-genomic analysis of new samples. These associations have been previously described as leading to fascinating genomic evolution in the symbiont (McCutcheon and Moran 2012). The bacterial genomes exhibit a significant reduction in size and the range of functions performed. They typically lose the ability to produce many metabolites or biobricks created by the host, and instead, streamline their metabolism by focusing on the amino acids that the host cannot produce. This level of co-evolution suggests a stable association between the two partners. However, the new data suggests a much more complex pattern as multiple independent acquisitions of co-symbionts are observed. Co-symbiont acquisition leads to a partition of the functions carried out on the bacterial side, with the new co-symbiont taking over some of the functions previously performed by Buchnera. In most cases, the new co-symbiont also brings the ability to produce B1 vitamin. Various facultative symbiotic taxa are recruited to be co-symbionts, with the frequency of acquisition related to the bacterial niche and lifestyle. REFERENCES Manzano-Marín, Alejandro, Armelle Coeur D’acier, Anne-Laure Clamens, Corinne Cruaud, Valérie Barbe, and Emmanuelle Jousselin. 2023. “Co-Obligate Symbioses Have Repeatedly Evolved across Aphids, but Partner Identity and Nutritional Contributions Vary across Lineages.” bioRxiv, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.08.28.505559. McCutcheon, John P., and Nancy A. Moran. 2012. “Extreme Genome Reduction in Symbiotic Bacteria.” Nature Reviews Microbiology 10 (1): 13–26. https://doi.org/10.1038/nrmicro2670. | Co-obligate symbioses have repeatedly evolved across aphids, but partner identity and nutritional contributions vary across lineages | Alejandro Manzano-Marín, Armelle Coeur d'acier, Anne-Laure Clamens, Corinne Cruaud, Valérie Barbe, Emmanuelle Jousselin | <p style="text-align: justify;">Aphids are a large family of phloem-sap feeders. They typically rely on a single bacterial endosymbiont, <em>Buchnera aphidicola</em>, to supply them with essential nutrients lacking in their diet. This association ... | | Genome Evolution, Other, Species interactions | Olivier Tenaillon | 2022-11-16 10:13:37 | ||
11 Oct 2021

Landscape connectivity alters the evolution of density-dependent dispersal during pushed range expansionsPhenotypic evolution during range expansions is contingent on connectivity and density dependenceRecommended by Inês Fragata based on reviews by 3 anonymous reviewersUnderstanding the mechanisms underlying range expansions is key for predicting species distributions in response to environmental changes (such as global warming) and managing the global expansion of invasive species (Parmesan 2006; Suarez & Tsutsui 2008). Traditionally, two types of ecological processes were studied as essential in shaping range expansion: dispersal and population growth. However, ecology and evolution are intertwined in range expansions, as phenotypic evolution of traits involved in demographic and dispersal patterns and processes can affect and be affected by ecological dynamics, representing a full eco-evolutionary loop (Williams et al. 2019; Miller et al. 2020). Range expansions can be characterized by the type of population growth and dispersal, divided into pushed or pulled range expansions. Species that have high dispersal and high population growth at low densities present pulled range expansions (pulled by individuals from the edge populations). In contrast, populations presenting increased growth rate at intermediate densities (due to Allee effects - Allee & Bowen 1932; i.e. where growth rate decreases at lower densities) and high dispersal at high densities present pushed range expansions (driven by individuals from core and intermediate populations) (Gandhi et al. 2016). Importantly, the type of expansion is expected to have very different consequences on the genetic (and therefore) phenotypic composition of core and edge populations. Specifically, genetic variability is expected to be lower in populations experiencing pulled expansions and higher in populations involved in pushed expansions (Gandhi et al. 2016; Miller et al. 2020). However, it is not always possible to distinguish between pulled and pushed expansions, as variation in speed and shape can overlap between the two types. In addition, it is difficult to experimentally manipulate the strength of the Allee effect to create pushed versus pulled expansions. Thus, several critical predictions regarding the genetic and phenotypic composition of pulled and pushed expansions are lacking empirical tests (but see Gandhi et al. 2016). In a previous study, Dahirel et al. (2021a) combined simulations and experimental evolution of the small wasps Trichogramma brassicae to show that low connectivity led to more pushed expansions, and higher connectivity generated more pulled expansions. In accordance with theoretical predictions, this led to reduced genetic diversity in pulled expansions, and the reverse pattern in pushed expansions. However, the question of how pulled and pushed expansions affect trait evolution remained unanswered. In this follow-up study, Dahirel et al. (2021b) tackled this issue and linked the changes in connectivity and type of expansion with the phenotypic evolution of several traits using individuals from their previous experiment. Namely, the authors compared core and edge populations with founder strains to test how evolution in pushed vs. pulled expansions affected wasp size, short movement, fecundity, dispersal, and density dependent dispersal. When density dependence was not accounted for, phenotypic changes in edge populations did not match the expectations from changes in expansion dynamics. This could be due to genetic trade-offs between traits that limit phenotypic evolution (Urquhart & Williams 2021). However, when accounting for density dependent dispersal, Dahirel et al. (2021b) observed that more connected landscapes (with pulled expansions) showed positive density dispersal in core populations and negative density dispersal in edge populations, similarly to other studies (e.g. Fronhofer et al. 2017). Interestingly, in pushed (with lower connectivity) landscapes, such shift was not observed. Instead, edge populations maintained positive density dispersal even after 14 generations of expansion, whereas core populations showed higher dispersal at lower density. The authors suggest that this seemingly contradictory result is due to a combination of three processes: 1) the expansion reduced positive density dispersal in edge populations; 2) reduced connectivity directly increased dispersal costs, increasing high density dispersal; and 3) reduced connectivity indirectly caused demographic stochasticity (and reduced temporal variability in patches) leading to higher dispersal at low density in core populations. However, these results must be taken with a grain of salt, since only one of the four experimental replicates were used in the density dependent dispersal experiment. In range expansions experiments, replication is fundamental, since stochastic processes (such as gene surfing, where alleles maybe rise in frequency due by chance) are prevalent (Miller et al. 2020), and results are highly dependent on sample size, or number of replicate populations analysed. Having said that, results from Dahirel et al. (2021b) highlight the importance to contextualize the management of invasions and species distribution, since it is thought that pulled expansions are more prevalent in nature, but pushed expansions can be more important in scenarios where patchiness is high, such as urban landscapes. Moreover, Dahirel's et al. (2021b) study is a first step showing that accounting for trait density dependence is crucial when following phenotypic evolution during range expansion, and that evolution of density dependent traits may be constrained by landscape conditions. This highlights the need to account for both connectivity and density dependence to draw more accurate predictions on the evolutionary and ecological outcomes of range expansions. Allee WC, Bowen ES (1932) Studies in animal aggregations: Mass protection against colloidal silver among goldfishes. Journal of Experimental Zoology, 61, 185–207. https://doi.org/10.1002/jez.1400610202 Dahirel M, Bertin A, Calcagno V, Duraj C, Fellous S, Groussier G, Lombaert E, Mailleret L, Marchand A, Vercken E (2021a) Landscape connectivity alters the evolution of density-dependent dispersal during pushed range expansions. bioRxiv, 2021.03.03.433752, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.03.03.433752 Dahirel M, Bertin A, Haond M, Blin A, Lombaert E, Calcagno V, Fellous S, Mailleret L, Malausa T, Vercken E (2021b) Shifts from pulled to pushed range expansions caused by reduction of landscape connectivity. Oikos, 130, 708–724. https://doi.org/10.1111/oik.08278 Fronhofer EA, Gut S, Altermatt F (2017) Evolution of density-dependent movement during experimental range expansions. Journal of Evolutionary Biology, 30, 2165–2176. https://doi.org/10.1111/jeb.13182 Gandhi SR, Yurtsev EA, Korolev KS, Gore J (2016) Range expansions transition from pulled to pushed waves as growth becomes more cooperative in an experimental microbial population. Proceedings of the National Academy of Sciences, 113, 6922–6927. https://doi.org/10.1073/pnas.1521056113 Miller TEX, Angert AL, Brown CD, Lee-Yaw JA, Lewis M, Lutscher F, Marculis NG, Melbourne BA, Shaw AK, Szűcs M, Tabares O, Usui T, Weiss-Lehman C, Williams JL (2020) Eco-evolutionary dynamics of range expansion. Ecology, 101, e03139. https://doi.org/10.1002/ecy.3139 Parmesan C (2006) Ecological and Evolutionary Responses to Recent Climate Change. Annual Review of Ecology, Evolution, and Systematics, 37, 637–669. https://doi.org/10.1146/annurev.ecolsys.37.091305.110100 Suarez AV, Tsutsui ND (2008) The evolutionary consequences of biological invasions. Molecular Ecology, 17, 351–360. https://doi.org/10.1111/j.1365-294X.2007.03456.x Urquhart CA, Williams JL (2021) Trait correlations and landscape fragmentation jointly alter expansion speed via evolution at the leading edge in simulated range expansions. Theoretical Ecology. https://doi.org/10.1007/s12080-021-00503-z Williams JL, Hufbauer RA, Miller TEX (2019) How Evolution Modifies the Variability of Range Expansion. Trends in Ecology & Evolution, 34, 903–913. https://doi.org/10.1016/j.tree.2019.05.012 | Landscape connectivity alters the evolution of density-dependent dispersal during pushed range expansions | Maxime Dahirel, Aline Bertin, Vincent Calcagno, Camille Duraj, Simon Fellous, Géraldine Groussier, Eric Lombaert, Ludovic Mailleret, Anaël Marchand, Elodie Vercken | <p style="text-align: justify;">As human influence reshapes communities worldwide, many species expand or shift their ranges as a result, with extensive consequences across levels of biological organization. Range expansions can be ranked on a con... | | Evolutionary Ecology, Experimental Evolution | Inês Fragata | 2021-03-05 17:15:46 | ||
01 Mar 2024
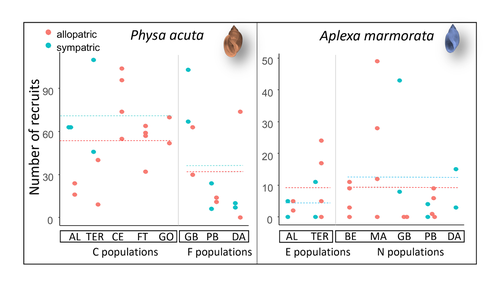
Rapid life-history evolution reinforces competitive asymmetry between invasive and resident speciesThe evolution of a hobo snailRecommended by Ben Phillips based on reviews by David Reznick and 2 anonymous reviewersAt the very end of a paper entitled "Copepodology for the ornithologist" Hutchinson (1951) pointed out the possibility of 'fugitive species'. A fugitive species, said Hutchinson, is one that we would typically think of as competitively inferior. Wherever it happens to live it will eventually be overwhelmed by competition from another species. We would expect it to rapidly go extinct but for one reason: it happens to be a much better coloniser than the other species. Now all we need to explain its persistence is a dose of space and a little disturbance: a world in which there are occasional disturbances that cause local extinction of the dominant species. Now, argued Hutchinson, we have a recipe for persistence, albeit of a harried kind. As Hutchinson put it, fugitive species "are forever on the move, always becoming extinct in one locality as they succumb to competition, and always surviving as they reestablish themselves in some other locality." It is a fascinating idea, not just because it points to an interesting strategy, but also because it enriches our idea of competition: competition for space can be just as important as competition for time. Hutchinson's idea was independently discovered with the advent of metapopulation theory (Levins 1971; Slatkin 1974) and since then, of course, ecologists have gone looking, and they have unearthed many examples of species that could be said to have a fugitive lifestyle. These fugitive species are out there, but we don't often get to see them evolve. In their recent paper, Chapuis et al. (2024) make a convincing case that they have seen the evolution of a fugitive species. They catalog the arrival of an invasive freshwater snail on Guadeloupe in the Lesser Antilles, and they wonder what impact this snail's arrival might have on a native freshwater snail. This is a snail invasion, so it has been proceeding at a majestic pace, allowing the researchers to compare populations of the native snail that are completely naive to the invader with those that have been exposed to the invader for either a relatively short period (<20 generations) or longer periods (>20 generations). They undertook an extensive set of competition assays on these snails to find out which species were competitively superior and how the native species' competitive ability has evolved over time. Against naive populations of the native, the invasive snail turns out to be unequivocally the stronger competitor. (This makes sense; it probably wouldn't have been able to invade if it wasn't.) So what about populations of the native snail that have been exposed for longer, that have had time to adapt? Surprisingly these populations appear to have evolved to become even weaker competitors than they already were. So why is it that the native species has not simply been driven extinct? Drawing on their previous work on this system, the authors can explain this situation. The native species appears to be the better coloniser of new habitats. Thus, it appears that the arrival of the invasive species has pushed the native species into a different place along the competition-colonisation axis. It has sacrificed competitive ability in favour of becoming a better coloniser; it has become a fugitive species in its own backyard. This is a really nice empirical study. It is a large lab study, but one that makes careful sampling around a dynamic field situation. Thus, it is a lab study that informs an earlier body of fieldwork and so reveals a fascinating story about what is happening in the field. We are left not only with a particularly compelling example of character displacement towards a colonising phenotype but also with something a little less scientific: the image of a hobo snail, forever on the run, surviving in the spaces in between. References Chapuis E, Jarne P, David P (2024) Rapid life-history evolution reinforces competitive asymmetry between invasive and resident species. bioRxiv, 2023.10.25.563987, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.10.25.563987 Hutchinson, G.E. (1951) Copepodology for the Ornithologist. Ecology 32: 571–77. https://doi.org/10.2307/1931746 Levins, R., and D. Culver. (1971) Regional Coexistence of Species and Competition between Rare Species. Proceedings of the National Academy of Sciences 68, no. 6: 1246–48. https://doi.org/10.1073/pnas.68.6.1246. Slatkin, Montgomery. (1974) Competition and Regional Coexistence. Ecology 55, no. 1: 128–34. https://doi.org/10.2307/1934625. | Rapid life-history evolution reinforces competitive asymmetry between invasive and resident species | Elodie Chapuis, Philippe Jarne, Patrice David | <p style="text-align: justify;">Biological invasions by phylogenetically and ecologically similar competitors pose an evolutionary challenge to native species. Cases of character displacement following invasions suggest that they can respond to th... | | Evolutionary Ecology, Life History, Species interactions | Ben Phillips | 2023-10-26 15:49:33 | ||
16 Dec 2022
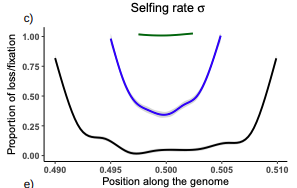
Conditions for maintaining and eroding pseudo-overdominance and its contribution to inbreeding depressionPseudo-overdominance: how linkage and selection can interact and oppose to purging of deleterious mutations.Recommended by Sylvain Glémin based on reviews by Yaniv Brandvain, Lei Zhao and 1 anonymous reviewerMost mutations affecting fitness are deleterious and they have many evolutionary consequences. The dynamics and consequences of deleterious mutations are a long-standing question in evolutionary biology and a strong theoretical background has already been developed, for example, to predict the mutation load, inbreeding depression or background selection. One of the classical results is that inbreeding helps purge partially recessive deleterious mutations by exposing them to selection in homozygotes. However, this mainly results from single-locus considerations. When interactions among several, more or less linked, deleterious mutations are taken into account, peculiar dynamics can emerge. One of them, called pseudo-overdominance (POD), corresponds to the maintenance in a population of two (or more) haplotype blocks composed of several recessive deleterious mutations in repulsion that mimics overdominance. Indeed, homozygote individuals for one of the haplotype blocks expose many deleterious mutations to selection whereas they are reciprocally masked in heterozygotes, leading to higher fitness of heterozygotes compared to both homozygotes. A related process, called associative overdominance (AOD) is the effect of such deleterious alleles in repulsion on the linked neutral variation that can be increased by AOD. Although this possibility has been recognized for a long time (Otha and Kimura 1969), it has been mainly considered an anecdotal process. Recently, both theoretical (Zhao and Charlesworth 2016) and genomic analyses (Gilbert et al. 2020) have renewed interest in such a process, suggesting that it could be important in weakly recombining regions of a genome. Donald Waller (2021) - one of the co-authors of the current work - also recently proposed that POD could be quantitatively important with broad implications, and could resolve some unexplained observations such as the maintenance of inbreeding depression in highly selfing species. Yet, a proper theoretical framework analysing the effect of inbreeding on POD was lacking. In this theoretical work, Diala Abu Awad and Donald Waller (2022) addressed this question through an elegant combination of analytical predictions and intensive multilocus simulations. They determined the conditions under which POD can be maintained and how long it could resist erosion by recombination, which removes the negative association between deleterious alleles (repulsion) at the core of the mechanism. They showed that under tight linkage, POD regions can persist for a long time and generate substantial segregating load and inbreeding depression, even under inbreeding, so opposing (for a while) to the purging effect. They also showed that background selection can affect the genomic structure of POD regions by rapidly erasing weak POD regions but maintaining strong POD regions (i.e with many tightly linked deleterious alleles). These results have several implications. They can explain the maintenance of inbreeding depression despite inbreeding (as anticipated by Waller 2021), which has implications for the evolution of mating systems. If POD can hardly emerge under high selfing, it can persist from an outcrossing ancestor long after the transition towards a higher selfing rate and could explain the maintenance of mixed mating systems(which is possible with true overdominance, see Uyenoyama and Waller 1991). The results also have implications for genomic analyses, pointing to regions of low or no recombination where POD could be maintained, generating both higher diversity and heterozygosity than expected and variance in fitness. As structural variations are likely widespread in genomes with possible effects on suppressing recombination (Mérot et al. 2020), POD regions should be checked more carefully in genomic analyses (see also Gilbert et al. 2020). Overall, this work should stimulate new theoretical and empirical studies, especially to assess how quantitatively strong and widespread POD can be. It also stresses the importance of properly considering genetic linkage genome-wide, and so the role of recombination landscapes in determining patterns of diversity and fitness effects. References
Awad DA, Waller D (2022) Conditions for maintaining and eroding pseudo-overdominance and its contribution to inbreeding depression. bioRxiv, 2021.12.16.473022, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.12.16.473022 Gilbert KJ, Pouyet F, Excoffier L, Peischl S (2020) Transition from Background Selection to Associative Overdominance Promotes Diversity in Regions of Low Recombination. Current Biology, 30, 101-107.e3. https://doi.org/10.1016/j.cub.2019.11.063 Mérot C, Oomen RA, Tigano A, Wellenreuther M (2020) A Roadmap for Understanding the Evolutionary Significance of Structural Genomic Variation. Trends in Ecology & Evolution, 35, 561–572. https://doi.org/10.1016/j.tree.2020.03.002 Ohta T, Kimura M (1969) Linkage disequilibrium at steady state determined by random genetic drift and recurrent mutation. Genetics, 63, 229–238. https://doi.org/10.1093/genetics/63.1.229 Uyenoyama MK, Waller DM (1991) Coevolution of self-fertilization and inbreeding depression II. Symmetric overdominance in viability. Theoretical Population Biology, 40, 47–77. https://doi.org/10.1016/0040-5809(91)90046-I Waller DM (2021) Addressing Darwin’s dilemma: Can pseudo-overdominance explain persistent inbreeding depression and load? Evolution, 75, 779–793. https://doi.org/10.1111/evo.14189 Zhao L, Charlesworth B (2016) Resolving the Conflict Between Associative Overdominance and Background Selection. Genetics, 203, 1315–1334. https://doi.org/10.1534/genetics.116.188912 | Conditions for maintaining and eroding pseudo-overdominance and its contribution to inbreeding depression | Diala Abu Awad, Donald Waller | <p style="text-align: justify;">Classical models that ignore linkage predict that deleterious recessive mutations should purge or fix within inbred populations, yet inbred populations often retain moderate to high segregating load. True overdomina... | | Evolutionary Dynamics, Evolutionary Theory, Genome Evolution, Hybridization / Introgression, Population Genetics / Genomics, Reproduction and Sex | Sylvain Glémin | 2022-01-04 12:15:35 | ||
13 Nov 2023
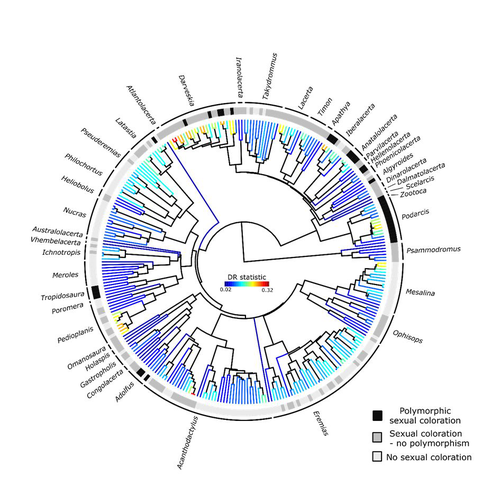
Color polymorphism and conspicuousness do not increase speciation rates in LacertidsColour polymorphism does not increase diversification rates in lizardsRecommended by Alejandro Gonzalez Voyer based on reviews by 2 anonymous reviewersThe striking differences in species richness among lineages in the Tree of Life have long attracted much research interest. In particular, researchers have asked whether certain traits are associated with greater diversification, with a particular focus on traits under sexual selection given their direct link to mating isolation. Polymorphism, defined as the presence of co-occurring, heritable morphs within a population, has been proposed to influence diversification rates although the effect has been proposed as both promoting or alternatively impeding speciation. The effect of polymorphism may be positive, that is facilitating speciation if polymorphism allows to broaden the ecological niche, thus enabling range expansion, or enabling maintenance of populations in variable environments. Specialized ectomorphs have been observed in several species (e.g. Kusche et al. 2015, Lattanzio and Miles 2016, Whitney et al. 2018, Scali et al. 2016). Polymorphism may also facilitate speciation if a morph is lost during the colonization of a novel area or niche, resulting in rapid divergence of the remaining morphs and reproductive isolation from the ancestral population, known as the morph speciation hypothesis (West-Eberhard 1986, Corl et al. 2010). On the other hand, polymorphism may hamper speciation through disassortative maintaining by morph, which may maintain the polymorphism through the speciation process (Jamie and Meier 2020). An example of such a process is Heliconius numata where disassortative mate preferences based on color hampers ecological speciation (Chouteau et al. 2017). Previous evidence in birds and lizards suggests polymorphism favors diversification (Corl et al. 2010b, 2012, Hugall and Stuart-Fox 2012, Brock et al. 2021). Here, de Solan et al. (2023) test the effect of polymorphism on diversification in Lacertidae, a family of lizards containing more than 300 species distributed across Europe, Africa and Asia. The group offers a good model system to test the effect of polymorphism on speciation as it contains several species with colour polymorphism, sometimes present in both sexes but restricted to males when present in the flank. Using coloration data from the literature as well as photographs of live specimens for 295 species the authors tested whether the presence of polymorphism is associated with higher diversification rates. While undertaking their project, another group independently tackled the same question (Brock et al. 2021), using the same model system but coming to very different conclusions. Therefore, de Solan et al. (2023) decided to also contrast their results with those of Brock et al. (2021) to determine the factors responsible for the contrasting results of both studies. The latter I consider one of the strengths of the work, given the careful re-analyses to determine the causes of the discrepancies between both studies. De Solan et al. (2023) found no association between the presence of polymorphism and diversification rates, even though they used different analytical approaches. Thus, this study is interesting as it provides results that do not support a positive effect of polymorphism on species richness. The use of a phylogeny with more limited species sampling (García-Porta et al. 2019) implied that the authors had to manually add 75 species, of which 17 were added to the tree based on information from previously published trees and 68 were added at random locations within the genus. To control for potential biases the authors repeated the analyses using a sample of trees with the imputed taxa, results were broadly concordant across the set of trees. The careful re-analysis contrasting Brock et al. (2021) and de Solan et al. (2023) results suggests the difference is mainly due to a difference in how species were coded as presenting polymorphism, which differed between the two studies, as well as a difference in the package version used to run the state-dependent diversification models. Interestingly non-parametric analyses yielded similar results across both datasets. Garcia-Porta, J., Irisarri, I., Kirchner, M. et al. 2019. Environmental temperatures shape thermal physiology as well as diversification and genome-wide substitution rates in lizards. Nature Communications. 10: 4077. https://doi.org/10.1038/s41467-019-11943-x de Solan T, Sinervo B, Geniez P, David P, Crochet P-A (2023) Colour polymorphism and conspicuousness do not increase speciation rates in Lacertids. bioRxiv, 2023.02.15.528678, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.02.15.528678 West-Eberhard, M.J. 1986. Alternative adaptations, speciation, and phylogeny (A review). Proceedings of the National Academy of Sciences. 83: 1388-1392. https://doi.org/10.1073/pnas.83.5.1388 | Color polymorphism and conspicuousness do not increase speciation rates in Lacertids | Thomas de Solan, Barry Sinervo, Philippe Geniez, Patrice David, Pierre-André Crochet | <p style="text-align: justify;">Conspicuous body colors and color polymorphism have been hypothesized to increase rates of speciation. Conspicuous colors are evolutionary labile, and often involved in intraspecific sexual signaling and thus may pr... | | Evolutionary Ecology, Macroevolution, Speciation | Alejandro Gonzalez Voyer | 2023-02-22 10:05:03 |




