
Latest recommendations

| Id▲ | Title * | Authors * | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
17 Nov 2017

ABC random forests for Bayesian parameter inferenceMachine learning methods are useful for Approximate Bayesian Computation in evolution and ecologyRecommended by Michael Blum based on reviews by Dennis Prangle and Michael BlumIt is my pleasure to recommend the paper by Raynal et al. [1] about using random forest for parameter inference. There are two reviews about the paper, one review written by Dennis Prangle and another review written by myself. Both reviews were positive and included comments that have been addressed in the current version of the preprint. The paper nicely shows that modern machine learning approaches are useful for Approximate Bayesian Computation (ABC) and more generally for simulation-driven parameter inference in ecology and evolution. The authors propose to consider the random forest approach, proposed by Meinshausen [2] to perform quantile regression. The numerical implementation of ABC with random forest, available in the abcrf package, is based on the RANGER R package that provides a fast implementation of random forest for high-dimensional data. According to my reading of the manuscript, there are 3 main advantages when using random forest (RF) for parameter inference with ABC. The first advantage is that RF can handle many summary statistics and that dimension reduction is not needed when using RF. The second advantage is very nicely displayed in Figure 5, which shows the main result of the paper. If correct, 95% posterior credibility intervals (C.I.) should contain 95% of the parameter values used in simulations. Figure 5 shows that posterior C.I. obtained with rejection are too large compared to other methods. By contrast, C.I. obtained with regression methods have been shrunken. However, the shrinkage can be excessive for the smallest tolerance rates, with coverage values that can be equal to 85% instead of the expected 95% value. The attractive property of RF is that C.I. have been shrunken but the coverage is of 100% resulting in a conservative decision about parameter values. The last advantage is that no hyperparameter should be chosen. It is a parameter free approach, which is desirable because of the potential difficulty of choosing an appropriate acceptance rate. The main drawback of the proposed approach concerns joint parameter inference. There are many settings where the joint parameter distribution is of interest and the proposed RF approach cannot handle that. In population genetics for example, estimation of the severity and of the duration of the bottleneck should be estimated jointly because of identifiability issues. The challenge of performing joint parameter inference with RF might constitute a useful research perspective. References [1] Raynal L, Marin J-M, Pudlo P, Ribatet M, Robert CP, Estoup A. 2017. ABC random forests for Bayesian parameter inference. arXiv 1605.05537v4, https://arxiv.org/pdf/1605.05537 | ABC random forests for Bayesian parameter inference | Louis Raynal, Jean-Michel Marin, Pierre Pudlo, Mathieu Ribatet, Christian P. Robert, Arnaud Estoup | This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (http:// dx.doi.org/ 10.24072/ pci.evolbiol.100036). Approximate Bayesian computation (ABC) has grown into a standard methodology that manages Bayesian infer... | | Bioinformatics & Computational Biology, Evolutionary Applications, Other, Population Genetics / Genomics | Michael Blum | 2017-07-06 07:42:00 | ||
07 Sep 2018

Parallel pattern of differentiation at a genomic island shared between clinal and mosaic hybrid zones in a complex of cryptic seahorse lineagesGenomic parallelism in adaptation to orthogonal environments in sea horsesRecommended by Yaniv Brandvain based on reviews by 2 anonymous reviewersStudies in speciation genomics have revealed that gene flow is quite common, and that despite this, species can maintain their distinct environmental adaptations. Although researchers are still elucidating the genomic mechanisms by which species maintain their adaptations in the face of gene flow, this often appears to involve few diverged genomic regions in otherwise largely undifferentiated genomes. In this preprint [1], Riquet and colleagues investigate the genetic structuring and patterns of parallel evolution in the long-snouted seahorse. References [1] Riquet, F., Liautard-Haag, C., Woodall, L., Bouza, C., Louisy, P., Hamer, B., Otero-Ferrer, F., Aublanc, P., Béduneau, V., Briard, O., El Ayari, T., Hochscheid, S. Belkhir, K., Arnaud-Haond, S., Gagnaire, P.-A., Bierne, N. (2018). Parallel pattern of differentiation at a genomic island shared between clinal and mosaic hybrid zones in a complex of cryptic seahorse lineages. bioRxiv, 161786, ver. 4 recommended and peer-reviewed by PCI Evol Biol. doi: 10.1101/161786 | Parallel pattern of differentiation at a genomic island shared between clinal and mosaic hybrid zones in a complex of cryptic seahorse lineages | Florentine Riquet, Cathy Liautard-Haag, Lucy Woodall, Carmen Bouza, Patrick Louisy, Bojan Hamer, Francisco Otero-Ferrer, Philippe Aublanc, Vickie Béduneau, Olivier Briard, Tahani El Ayari, Sandra Hochscheid, Khalid Belkhir, Sophie Arnaud-Haond, Pi... | <p>Diverging semi-isolated lineages either meet in narrow clinal hybrid zones, or have a mosaic distribution associated with environmental variation. Intrinsic reproductive isolation is often emphasized in the former and local adaptation in the la... | | Hybridization / Introgression, Molecular Evolution, Population Genetics / Genomics, Speciation | Yaniv Brandvain | Kathleen Lotterhos, Sarah Fitzpatrick | 2017-07-11 13:12:40 | |
21 Nov 2018
Convergent evolution as an indicator for selection during acute HIV-1 infectionIs convergence an evidence for positive selection?Recommended by Guillaume Achaz based on reviews by Jeffrey Townsend and 1 anonymous reviewerThe preprint by Bertels et al. [1] reports an interesting application of the well-accepted idea that positively selected traits (here variants) can appear several times independently; think about the textbook examples of flight capacity. Hence, the authors assume that reciprocally convergence implies positive selection. The methodology becomes then, in principle, straightforward as one can simply count variants in independent datasets to detect convergent mutations. References [1] Bertels, F., Metzner, K. J., & Regoes R. R. (2018). Convergent evolution as an indicator for selection during acute HIV-1 infection. BioRxiv, 168260, ver. 4 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/168260 | Convergent evolution as an indicator for selection during acute HIV-1 infection | Frederic Bertels, Karin J Metzner, Roland R Regoes | <p>Convergent evolution describes the process of different populations acquiring similar phenotypes or genotypes. Complex organisms with large genomes only rarely and only under very strong selection converge to the same genotype. In contrast, ind... | | Bioinformatics & Computational Biology, Evolutionary Applications, Genome Evolution, Molecular Evolution | Guillaume Achaz | 2017-07-26 08:39:17 | ||
28 Feb 2018

Insects and incest: sib-mating tolerance in natural populations of a parasitoid waspIncestuous insects in nature despite occasional fitness costsRecommended by Caroline Nieberding and Bertanne Visser based on reviews by 2 anonymous reviewersInbreeding, or mating between relatives, generally lowers fitness [1]. Mating between genetically similar individuals can result in higher levels of homozygosity and consequently a higher frequency with which recessive disease alleles may be expressed within a population. Reduced fitness as a consequence of inbreeding, or inbreeding depression, can vary between individuals, sexes, populations and species [2], but remains a pervasive challenge for many organisms with small local population sizes, including humans [3]. But all is not lost for individuals within small populations, because an array of mechanisms can be employed to evade the negative effects of inbreeding [4], including sib-mating avoidance and dispersal [5, 6]. Despite thorough investigation of inbreeding and sib-mating avoidance in the laboratory, only very few studies have ventured into the field besides studies on vertebrates and eusocial insects. The study of Collet et al. [7] is a surprising exception, where the effect of male density and frequency of relatives on inbreeding avoidance was tested in the laboratory, after which robust field collections and microsatellite genotyping were used to infer relatedness and dispersal in natural populations. The parasitic wasp Venturia canescens is an excellent model system to study inbreeding, because mating success was previously found to decrease with increasing relatedness between mates in the laboratory [8] and this species thus suffers from inbreeding depression [9–11]. The authors used an elegant design combining population genetics and model simulations to estimate relatedness of mating partners in the field and compared that with a theoretical distribution of potential mate encounters when random mating is assumed. One of the most important findings of this study is that mating between siblings is not avoided in this species in the wild, despite negative fitness effects when inbreeding does occur. Similar findings were obtained for another insect species, the field cricket Gryllus campestris [12], which leaves us to wonder whether inbreeding tolerance could be more common in nature than currently appreciated. The authors further looked into sex-specific dispersal patterns between two patches located a few hundred meters apart. Females were indeed shown to be more related within a patch, but no genetic differences were observed between males, suggesting that V. canescens males more readily disperse. Moreover, microsatellite data at 18 different loci did not reveal genetic differentiation between populations approximately 300 kilometers apart. Gene flow is thus occurring over considerable distances, which could play an important role in the ability of this species to avoid negative fitness consequences of inbreeding in nature. Another interesting aspect of this work is that discrepancies were found between laboratory- and field-based data. What is the relevance of laboratory-based experiments if they cannot predict what is happening in the wild? Many, if not most, biologists (including us) bring our model system into the laboratory to control, at least to some extent, the plethora of environmental factors that could potentially affect our system (in ways that we do not want). Most behavioral studies on mating patterns and sexual selection are conducted in standardized laboratory conditions, but sexual selection is in essence social selection, because an individual’s fitness is partly determined by the phenotype of its social partners (i.e. the social environment) [13]. The social environment may actually dictate the expression of female mate choice and it is unclear how potential laboratory-induced social biases affect mating outcome. In V. canescens, findings using field-caught individuals paint a completely opposite picture of what was previously shown in the laboratory, i.e. sib-avoidance is not taking place in the field. It is likely that density, level of relatedness, sex ratio in the field, and/or the size of experimental arenas in the lab are all factors affecting mate selectivity, as we have previously shown in a butterfly [14–16]. If females, for example, typically only encounter a few males in sequence in the wild, it may be problematic for them to express choosiness when confronted simultaneously with two or more males in the laboratory. A recent study showed that, in the wild, female moths take advantage of staying in groups to blur male choosiness [17]. It is becoming more and more clear that what we observe in the laboratory may not actually reflect what is happening in nature [18]. Instead of ignoring the species-specific life history and ecological features of our favorite species when conducting lab experiments, we suggest that it is time to accept that we now have the theoretical foundations to tease apart what in this “environmental noise” actually shapes sexual selection in nature. Explicitly including ecology in studies on sexual selection will allow us to make more meaningful conclusions, i.e. rather than “this is what may happen in the wild”, we would be able to state “this is what often happens in nature”. References [1] Charlesworth D & Willis JH. 2009. The genetics of inbreeding depression. Nat. Rev. Genet. 10: 783–796. doi: 10.1038/nrg2664 | Insects and incest: sib-mating tolerance in natural populations of a parasitoid wasp | Marie Collet, Isabelle Amat, Sandrine Sauzet, Alexandra Auguste, Xavier Fauvergue, Laurence Mouton, Emmanuel Desouhant | <p>This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (http://dx.doi.org/10.24072/pci.evolbiol.100047) 1. Sib-mating avoidance is a pervasive behaviour that likely evolves in species subject to inbreeding dep... | | Behavior & Social Evolution, Evolutionary Ecology, Sexual Selection | Caroline Nieberding | 2017-07-28 09:23:20 | ||
03 Aug 2017
POSTPRINT
Fisher's geometrical model and the mutational patterns of antibiotic resistance across dose gradientsWhat doesn’t kill us makes us stronger: can Fisher’s Geometric model predict antibiotic resistance evolution?Recommended by Inês Fragata and Claudia Bank
The increasing number of reported cases of antibiotic resistance is one of today’s major public health concerns. Dealing with this threat involves understanding what drives the evolution of antibiotic resistance and investigating whether we can predict (and subsequently avoid or circumvent) it [1]. References [1] Palmer AC, and Kishony R. 2013. Understanding, predicting and manipulating the genotypic evolution of antibiotic resistance. Nature Review Genetics 14: 243—248. doi: 10.1038/nrg3351 [2] Tenaillon O. 2014. The utility of Fisher’s geometric model in evolutionary genetics. Annual Review of Ecology, Evolution and Systematics 45: 179—201. doi: 10.1146/annurev-ecolsys-120213-091846 [3] Blanquart F and Bataillon T. 2016. Epistasis and the structure of fitness landscapes: are experimental fitness landscapes compatible with Fisher’s geometric model? Genetics 203: 847—862. doi: 10.1534/genetics.115.182691 [4] Harmand N, Gallet R, Jabbour-Zahab R, Martin G and Lenormand T. 2017. Fisher’s geometrical model and the mutational patterns of antibiotic resistance across dose gradients. Evolution 71: 23—37. doi: 10.1111/evo.13111 [5] de Visser, JAGM, and Krug J. 2014. Empirical fitness landscapes and the predictability of evolution. Nature 15: 480—490. doi: 10.1038/nrg3744 [6] Palmer AC, Toprak E, Baym M, Kim S, Veres A, Bershtein S and Kishony R. 2015. Delayed commitment to evolutionary fate in antibiotic resistance fitness landscapes. Nature Communications 6: 1—8. doi: 10.1038/ncomms8385 | Fisher's geometrical model and the mutational patterns of antibiotic resistance across dose gradients | Noémie Harmand, Romain Gallet, Roula Jabbour-Zahab, Guillaume Martin, Thomas Lenormand | Fisher's geometrical model (FGM) has been widely used to depict the fitness effects of mutations. It is a general model with few underlying assumptions that gives a large and comprehensive view of adaptive processes. It is thus attractive in sever... | | Adaptation | Inês Fragata | 2017-08-01 16:06:02 | ||
20 Nov 2017
Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selectionUnderstanding genetic variance, load, and inbreeding depression with selfingRecommended by Aneil F. Agrawal based on reviews by Frédéric Guillaume and 1 anonymous reviewerA classic problem in evolutionary biology is to understand the genetic variance in fitness. The simplest hypothesis is that variation exists, even in well-adapted populations, as a result of the balance between mutational input and selective elimination. This variation causes a reduction in mean fitness, known as the mutation load. Though mutation load is difficult to quantify empirically, indirect evidence of segregating genetic variation in fitness is often readily obtained by comparing the fitness of inbred and outbred offspring, i.e., by measuring inbreeding depression. Mutation-selection balance models have been studied as a means of understanding the genetic variance in fitness, mutation load, and inbreeding depression. Since their inception, such models have increased in sophistication, allowing us to ask these questions under more realistic and varied scenarios. The new theoretical work by Abu Awad and Roze [1] is a substantial step forward in understanding how arbitrary levels of self-fertilization affect variation, load and inbreeding depression under mutation-selection balance. References [1] Abu Awad D and Roze D. 2017. Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selection. bioRxiv, 180000, ver. 4 of 17th November 2017. doi: 10.1101/180000 [2] Lande R. 1977. The influence of the mating system on the maintenance of genetic variability in polygenic characters. Genetics 86: 485–498. [3] Charlesworth D and Charlesworth B. 1987. Inbreeding depression and its evolutionary consequences. Annual Review of Ecology and Systematics. 18: 237–268. doi: 10.1111/10.1146/annurev.es.18.110187.001321 [4] Lande R and Porcher E. 2015. Maintenance of quantitative genetic variance under partial self-fertilization, with implications for the evolution of selfing. Genetics 200: 891–906. doi: 10.1534/genetics.115.176693 [5] Roze D. 2015. Effects of interference between selected loci on the mutation load, inbreeding depression, and heterosis. Genetics 201: 745–757. doi: 10.1534/genetics.115.178533 [6] Martin G and Lenormand T. 2006. A general multivariate extension of Fisher's geometrical model and the distribution of mutation fitness effects across species. Evolution 60: 893–907. doi: 10.1111/j.0014-3820.2006.tb01169.x [7] Martin G, Elena SF and Lenormand T. 2007. Distributions of epistasis in microbes fit predictions from a fitness landscape model. Nature Genetics 39: 555–560. doi: 10.1038/ng1998 | Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selection | Diala Abu Awad and Denis Roze | The mating system of a species is expected to have important effects on its genetic diversity. In this paper, we explore the effects of partial selfing on the equilibrium genetic variance Vg, mutation load L and inbreeding depression δ under stabi... | | Evolutionary Theory, Population Genetics / Genomics, Quantitative Genetics, Reproduction and Sex | Aneil F. Agrawal | 2017-08-26 09:29:20 | ||
11 Sep 2017

POSTPRINT
Less effective selection leads to larger genomesColonisation of subterranean ecosystems leads to larger genome in waterlouse (Aselloidea)Recommended by Benoit Nabholz and Jochen B. W. WolfThe total amount of DNA utilized to store hereditary information varies immensely among eukaryotic organisms. Single copy genome sizes – disregarding differences due to ploidy - differ by more than three orders of magnitude ranging from a few million nucleotides (Mb) to hundreds of billions (Gb). With the ever-increasing availability of fully sequenced genomes we now know that most of the difference is due either to whole genome duplication or to variation in the abundance of repetitive elements. Regarding repetitive elements, the evolutionary forces underlying the large variation 'allowing' more or less elements in a genome remain largely elusive. A tentative correlation between an organism's complexity (however this may be adequately measured) and genome size, the so called C-value paradox [1], has long been dismissed. Studies testing for selection on secondary phenotypic effects associated with genome size (cell size, metabolic rates, nutrient availability) have yielded mixed results. Nonadaptive theories capitalizing on a role of deleterious insertion-deletion mutations and genetic drift as the main drivers have likewise received mixed support [2-3]. Overall, most evidence was derived from analyses across broad taxonomical scales [4-6]. Lefébure and colleagues [7] take a different approach. They confine their considerations to a homogeneous, restricted taxonomical group, isopod crustaceans of the superfamily Aselloidea. This taxonomic focus allows the authors to circumvent many of the confounding factors such as phylogenetic inertia, life history divergence and mutation rate variation that tend to trouble analyses across broad taxonomic timescales. Another important feature of the chosen system is the evolutionary independent transition of habitat use that has occurred at least 11 times. One group of species inhabits subterranean ecosystems (groundwater), another group thrives on surface water. Populations of the former live in low-energy habitats and are expected to be outnumbered by their surface dwelling relatives. Interestingly – and a precondition for the study - the groundwater species have significantly larger genomes (up to 137%). With this unique set-up, the authors are able to investigate the link between genome size and evolutionary forces related to a proxy of long-term population size by removing many of the confounding factors a priori. Upfront, we learn that the dN/dS ratio is higher in the groundwater species. This may either suggest prevalent positive selection or lower efficacy of purifying selection (relaxed constraint) in the group of species in which population sizes are expected to be low. Using a series of population genetic analyses the authors provide compelling evidence for the latter. Analyses are carefully conducted and include models for estimating the intensity and frequency of purifying and positive selection, the DoS (direction of selection) and α statistic. Next the authors also exclude the possibility that increased dN/dS of the subterranean groundwater species may be due to nonfunctionalization, which may result from the subterranean lifestyle. Overall, these analyses suggest relaxed constraint in smaller populations as the most plausible alternative to explain increased dN/dS ratios. In addition to the efficacy of selection, the authors estimate the timing of the ecological transition under the rationale that the amount of time a species may have been exposed to the subterranean habitat may reflect long term population sizes. To calibrate the 'colonization clock' they apply a neat trick based on the degree of degeneration of the opsin gene (as vision tends to get lost in these habitats). When finally testing which parameters may explain differences in genome size all factors – ecological status, selection efficiency as measured by dN/dS and colonization time - turned out to be significant predictors. Direct estimates of the short term effective population size Ne from polymorphism data, however, did not correlate with genome size. Ruling out the effect of other co-variates such as body size and growth rate the authors conclude that genome size was overall best predicted by long-term population size change upon habitat shift. In that the authors provide convincing evidence that the increase in genome size is linked to a decrease in long-term reduction of selection efficiency of subterranean species. Assuming a bias for insertion mutations over deletion mutations (which is usually the case in eukaryotes) this result is in agreement with the theory of mutational hazard [4-6]. This theory proposed by Michael Lynch postulates that the accumulation of non-functional DNA has a weak deleterious effect that can only be efficiently opposed by natural selection in species with high Ne. In conclusion, Lefébure and colleagues provide novel and welcome evidence supporting a 'neutralist' hypothesis of genome size evolution without the need to invoke an adaptive component. Methodologically, the study cautions against the common use of polymorphism-based estimates of Ne which are often obfuscated by transitory demographic change. Instead, alternative measures of selection efficacy linked to long-term population size may serve as better predictors of genome size. We hope that this study will stimulate additional work testing the link between Ne and genome size variation in other taxonomical groups [8-9]. Using genome sequences instead of the transcriptome approach applied here may concomitantly further our understanding of the molecular mechanisms underlying genome size change. References [1] Thomas, CA Jr. 1971. The genetic organization of chromosomes. Annual Review of Genetics 5: 237–256. doi: 10.1146/annurev.ge.05.120171.001321 [2] Ågren JA, Greiner S, Johnson MTJ, Wright SI. 2015. No evidence that sex and transposable elements drive genome size variation in evening primroses. Evolution 69: 1053–1062. doi: 10.1111/evo.12627 [3] Bast J, Schaefer I, Schwander T, Maraun M, Scheu S, Kraaijeveld K. 2016. No accumulation of transposable elements in asexual arthropods. Molecular Biology and Evolution 33: 697–706. doi: 10.1093/molbev/msv261 [4] Lynch M. 2007. The Origins of Genome Architecture. Sinauer Associates. [5] Lynch M, Bobay LM, Catania F, Gout JF, Rho M. 2011. The repatterning of eukaryotic genomes by random genetic drift. Annual Review of Genomics and Human Genetics 12: 347–366. doi: 10.1146/annurev-genom-082410-101412 [6] Lynch M, Conery JS. 2003. The origins of genome complexity. Science 302: 1401–1404. doi: 10.1126/science.1089370 [7] Lefébure T, Morvan C, Malard F, François C, Konecny-Dupré L, Guéguen L, Weiss-Gayet M, Seguin-Orlando A, Ermini L, Der Sarkissian C, Charrier NP, Eme D, Mermillod-Blondin F, Duret L, Vieira C, Orlando L, and Douady CJ. 2017. Less effective selection leads to larger genomes. Genome Research 27: 1016-1028. doi: 10.1101/gr.212589.116 [8] Lower SS, Johnston JS, Stanger-Hall KF, Hjelmen CE, Hanrahan SJ, Korunes K, Hall D. 2017. Genome size in North American fireflies: Substantial variation likely driven by neutral processes. Genome Biolology and Evolution 9: 1499–1512. doi: 10.1093/gbe/evx097 [9] Sessegolo C, Burlet N, Haudry A. 2016. Strong phylogenetic inertia on genome size and transposable element content among 26 species of flies. Biology Letters 12: 20160407. doi: 10.1098/rsbl.2016.0407 | Less effective selection leads to larger genomes | Tristan Lefébure, Claire Morvan, Florian Malard, Clémentine François, Lara Konecny-Dupré, Laurent Guéguen, Michèle Weiss-Gayet, Andaine Seguin-Orlando, Luca Ermini, Clio Der Sarkissian, N. Pierre Charrier, David Eme, Florian Mermillod-Blondin, Lau... | The evolutionary origin of the striking genome size variations found in eukaryotes remains enigmatic. The effective size of populations, by controlling selection efficacy, is expected to be a key parameter underlying genome size evolution. However... | | Evolutionary Theory, Genome Evolution, Molecular Evolution, Population Genetics / Genomics | Benoit Nabholz | 2017-09-08 09:39:23 | ||
19 Feb 2018

Genomic imprinting mediates dosage compensation in a young plant XY systemDosage compensation by upregulation of maternal X alleles in both males and females in young plant sex chromosomesRecommended by Tatiana Giraud and Judith Mank based on reviews by 3 anonymous reviewersSex chromosomes evolve as recombination is suppressed between the X and Y chromosomes. The loss of recombination on the sex-limited chromosome (the Y in mammals) leads to degeneration of both gene expression and gene content for many genes [1]. Loss of gene expression or content from the Y chromosome leads to differences in gene dose between males and females for X-linked genes. Because expression levels are often correlated with gene dose [2], these hemizygous genes have a lower expression levels in the heterogametic sex. This in turn disrupts the stoichiometric balance among genes in protein complexes that have components on both the sex chromosomes and autosomes [3], which could have serious deleterious consequences for the heterogametic sex. References | Genomic imprinting mediates dosage compensation in a young plant XY system | Aline Muyle, Niklaus Zemp, Cecile Fruchard, Radim Cegan, Jan Vrana, Clothilde Deschamps, Raquel Tavares, Franck Picard, Roman Hobza, Alex Widmer, Gabriel Marais | <p>During the evolution of sex chromosomes, the Y degenerates and its expression gets reduced relative to the X and autosomes. Various dosage compensation mechanisms that recover ancestral expression levels in males have been described in animals.... | | Bioinformatics & Computational Biology, Expression Studies, Genome Evolution, Molecular Evolution, Reproduction and Sex | Tatiana Giraud | 2017-09-20 20:39:46 | ||
19 Mar 2018

Natural selection on plasticity of thermal traits in a highly seasonal environmentIs thermal plasticity itself shaped by natural selection? An assessment with desert frogsRecommended by Wolf Blanckenhorn based on reviews by Dries Bonte, Wolf Blanckenhorn and Nadia Aubin-HorthIt is well known that climatic factors – most notably temperature, season length, insolation and humidity – shape the thermal niche of organisms on earth through the action of natural selection. But how is this achieved precisely? Much of thermal tolerance is actually mediated by phenotypic plasticity (as opposed to genetic adaptation). A prominent expectation is that environments with greater (daily and/or annual) thermal variability select for greater plasticity, i.e. better acclimation capacity. Thus, plasticity might be selected per se. A Chilean group around Leonardo Bacigalupe assessed natural selection in the wild in one marginal (and extreme) population of the four-eyed frog Pleurodema thaul (Anura: Leptodactylidae) in an isolated oasis in the Atacama Desert, permitting estimation of mortality without much potential of confounding it with migration [1]. Several thermal traits were considered: CTmax – the critical maximal temperature; CTmin – the critical minimum temperature; Tpref – preferred temperature; Q10 – thermal sensitivity of metabolism; and body mass. Animals were captured in the wild and subsequently assessed for thermal traits in the laboratory at two acclimation temperatures (10° & 20°C), defining the plasticity in all traits as the difference between the traits at the two acclimation temperatures. Thereafter the animals were released again in their natural habitat and their survival was monitored over the subsequent 1.5 years, covering two breeding seasons, to estimate viability selection in the wild. The authors found and conclude that, aside from larger body size increasing survival (an unsurprising result), plasticity does not seem to be systematically selected directly, while some of the individual traits show weak signs of selection. Despite limited sample size (ca. 80 frogs) investigated in only one marginal but very seasonal population, this study is interesting because selection on plasticity in physiological thermal traits, as opposed to selection on the thermal traits themselves, is rarely investigated. The study thus also addressed the old but important question of whether plasticity (i.e. CTmax-CTmin) is a trait by itself or an epiphenomenon defined by the actual traits (CTmax and CTmin) [2-5]. Given negative results, the main question could not be ultimately solved here, so more similar studies should be performed. References [1] Bacigalupe LD, Gaitan-Espitia, JD, Barria AM, Gonzalez-Mendez A, Ruiz-Aravena M, Trinder M & Sinervo B. 2018. Natural selection on plasticity of thermal traits in a highly seasonal environment. bioRxiv 191825, ver. 5 peer-reviewed by Peer Community In Evolutionary Biology. doi: 10.1101/191825 | Natural selection on plasticity of thermal traits in a highly seasonal environment | Leonardo Bacigalupe, Juan Diego Gaitan-Espitia, Aura M Barria, Avia Gonzalez-Mendez, Manuel Ruiz-Aravena, Mark Trinder, Barry Sinervo | <p>For ectothermic species with broad geographical distributions, latitudinal/altitudinal variation in environmental temperatures (averages and extremes) are expected to shape the evolution of physiological tolerances and the acclimation capacity ... | | Adaptation, Evolutionary Ecology, Phenotypic Plasticity | Wolf Blanckenhorn | 2017-09-22 23:17:40 | ||
09 Feb 2018
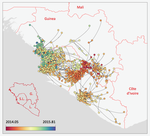
Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreakSimulating the effect of public health interventions using dated virus sequences and geographical dataRecommended by Samuel Alizon based on reviews by Christian Althaus, Chris Wymant and 1 anonymous reviewer
Perhaps because of its deadliness, the 2013-2016 Ebola Virus (EBOV) epidemics in West-Africa has led to unprecedented publication and sharing of full virus genome sequences. This was both rapid (90 full genomes were shared within weeks [1]) and important (more than 1500 full genomes have been released overall [2]). Furthermore, the availability of the metadata (especially GPS location) has led to depth analyses of the geographical spread of the epidemics [3]. References [1] Gire et al. 2014. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345: 1369–1372. doi: 10.1126/science.1259657. | Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreak | Simon Dellicour, Guy Baele, Gytis Dudas, Nuno R. Faria, Oliver G. Pybus, Marc A. Suchard, Andrew Rambaut, Philippe Lemey | <p>This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (https://doi.org/10.24072/pci.evolbiol.100046). The recent Ebola virus (EBOV) outbreak in West Africa witnessed considerable efforts to obtain viral genom... | | Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Samuel Alizon | 2017-09-30 13:49:57 |




