
Latest recommendations

| Id | Title | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers▲ | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
29 Sep 2022
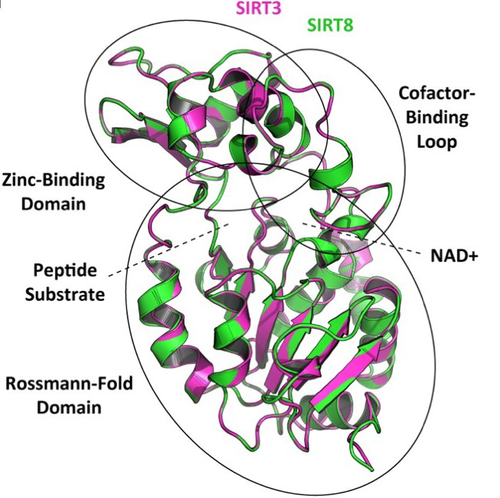
How many sirtuin genes are out there? evolution of sirtuin genes in vertebrates with a description of a new family memberMaking sense of vertebrate sirtuin genesRecommended by Frédéric Delsuc based on reviews by Filipe Castro, Nicolas Leurs and 1 anonymous reviewerSirtuin proteins are class III histone deacetylases that are involved in a variety of fundamental biological functions mostly related to aging. These proteins are located in different subcellular compartments and are associated with different biological functions such as metabolic regulation, stress response, and cell cycle control [1]. In mammals, the sirtuin gene family is composed of seven paralogs (SIRT1-7) grouped into four classes [2]. Due to their involvement in maintaining cell cycle integrity, sirtuins have been studied as a way to understand fundamental mechanisms governing longevity [1]. Indeed, the downregulation of sirtuin genes with aging seems to explain much of the pathophysiology that accumulates with aging [3]. Biomedical studies have thus explored the potential therapeutic implications of sirtuins [4] but whether they can effectively be used as molecular targets for the treatment of human diseases remains to be demonstrated [1]. Despite this biomedical interest and some phylogenetic analyses of sirtuin paralogs mostly conducted in mammals, a comprehensive evolutionary analysis of the sirtuin gene family at the scale of vertebrates was still lacking. In this preprint, Opazo and collaborators [5] took advantage of the increasing availability of whole-genome sequences for species representing all main groups of vertebrates to unravel the evolution of the sirtuin gene family. To do so, they undertook a phylogenomic approach in its original sense aimed at improving functional predictions by evolutionary analysis [6] in order to inventory the full vertebrate sirtuin gene repertoire and reconstruct its precise duplication history. Harvesting genomic databases, they extracted all predicted sirtuin proteins and performed phylogenetic analyses based on probabilistic inference methods. Maximum likelihood and Bayesian analyses resulted in well-resolved and congruent phylogenetic trees dividing vertebrate sirtuin genes into three major clades. These analyses also revealed an additional eighth paralog that was previously overlooked because of its restricted phyletic distribution. This newly identified sirtuin family member (named SIRT8) was recovered with unambiguous statistical support as a sister-group to the SIRT3 clade. Comparative genomic analyses based on conserved gene synteny confirmed that SIRT8 was present in all sampled non-amniote vertebrate genomes (cartilaginous fish, bony fish, coelacanth, lungfish, and amphibians) except cyclostomes. SIRT8 has thus most likely been lost in the last common ancestor of amniotes (mammals, reptiles, and birds). Discovery of such previously unknown genes in vertebrates is not completely surprising given the plethora of high-quality genomes now available. However, this study highlights the importance of considering a broad taxonomic sampling to infer evolutionary patterns of gene families that have been mostly studied in mammals because of their potential importance for human biology. Based on its phylogenetic position as closely related to SIRT3 within class I, it could be predicted that the newly identified SIRT8 paralog likely has a deacetylase activity and is probably located in mitochondria. To test these evolutionary predictions, Opazo and collaborators [5] conducted further bioinformatics analyses and functional experiments using the elephant shark (Callorhinchus milii) as a model species. RNAseq expression data were analyzed to determine tissue-specific transcription of sirtuin genes in vertebrates, including SIRT8 found to be mainly expressed in the ovary, which suggests a potential role in biological processes associated with reproduction. The elephant shark SIRT8 protein sequence was used with other vertebrates for comparative analyses of protein structure modeling and subcellular localization prediction both pointing to a probable mitochondrial localization. The protein localization and its function were further characterized by immunolocalization in transfected cells, and enzymatic and functional assays, which all confirmed the prediction that SIRT8 proteins are targeted to the mitochondria and have deacetylase activity. The extensive experimental efforts made in this study to shed light on the function of this newly discovered gene are both rare and highly commendable. Overall, this work by Opazo and collaborators [5] provides a comprehensive phylogenomic study of the sirtuin gene family in vertebrates based on detailed evolutionary analyses using state-of-the-art phylogenetic reconstruction methods. It also illustrates the power of adopting an integrative comparative approach supplementing the reconstruction of the duplication history of the gene family with complementary functional experiments in order to elucidate the function of the newly discovered SIRT8 family member. These results provide a reference phylogenetic framework for the evolution of sirtuin genes and the further functional characterization of the eight vertebrate paralogs with potential relevance for understanding the cellular biology of aging and its associated diseases in human. References [1] Vassilopoulos A, Fritz KS, Petersen DR, Gius D (2011) The human sirtuin family: Evolutionary divergences and functions. Human Genomics, 5, 485. https://doi.org/10.1186/1479-7364-5-5-485 [2] Yamamoto H, Schoonjans K, Auwerx J (2007) Sirtuin Functions in Health and Disease. Molecular Endocrinology, 21, 1745–1755. https://doi.org/10.1210/me.2007-0079 [3] Morris BJ (2013) Seven sirtuins for seven deadly diseases ofaging. Free Radical Biology and Medicine, 56, 133–171. https://doi.org/10.1016/j.freeradbiomed.2012.10.525 [4] Bordo D Structure and Evolution of Human Sirtuins. Current Drug Targets, 14, 662–665. http://dx.doi.org/10.2174/1389450111314060007 [5] Opazo JC, Vandewege MW, Hoffmann FG, Zavala K, Meléndez C, Luchsinger C, Cavieres VA, Vargas-Chacoff L, Morera FJ, Burgos PV, Tapia-Rojas C, Mardones GA (2022) How many sirtuin genes are out there? evolution of sirtuin genes in vertebrates with a description of a new family member. bioRxiv, 2020.07.17.209510, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.07.17.209510 [6] Eisen JA (1998) Phylogenomics: Improving Functional Predictions for Uncharacterized Genes by Evolutionary Analysis. Genome Research, 8, 163–167. https://doi.org/10.1101/gr.8.3.163 | How many sirtuin genes are out there? evolution of sirtuin genes in vertebrates with a description of a new family member | Juan C. Opazo, Michael W. Vandewege, Federico G. Hoffmann, Kattina Zavala, Catalina Meléndez, Charlotte Luchsinger, Viviana A. Cavieres, Luis Vargas-Chacoff, Francisco J. Morera, Patricia V. Burgos, Cheril Tapia-Rojas, Gonzalo A. Mardones | <p style="text-align: justify;">Studying the evolutionary history of gene families is a challenging and exciting task with a wide range of implications. In addition to exploring fundamental questions about the origin and evolution of genes, disent... | | Molecular Evolution | Frédéric Delsuc | Filipe Castro, Anonymous, Nicolas Leurs | 2022-05-12 16:06:04 | |
30 Jun 2023
How do monomorphic bacteria evolve? The Mycobacterium tuberculosis complex and the awkward population genetics of extreme clonalityHow the tubercle bacillus got its genome: modernising, modelling, and making sense of the stories we tellRecommended by B. Jesse Shapiro based on reviews by 2 anonymous reviewersIn this instructive review, Stritt and Gagneux offer a balanced perspective on the evolutionary forces shaping Mycobacterium tuberculosis and make the case that our instinct for storytelling be balanced with quantitative models. M. tuberculosis is perhaps the best-known clonal bacterial pathogen – evolving largely in the absence of horizontal gene transfer. Its genome is full of puzzling patterns, including much higher GC content than most intracellular pathogens (which suggests efficient selection to resist AT-skewed mutational bias) but a very high ratio of nonsynonymous to synonymous substitution rates (dN/dS ~ 0.5, typically interpreted as weak selection against deleterious amino acid changes). The authors offer alternative explanations for these patterns, framing the question: is M. tuberculosis evolution shaped mainly by drift or by efficient selection? They propose that this question can only be answered by accounting for the pathogen’s extreme clonality. A clonal lifestyle can have its advantages, for example when adaptations must arise in a particular order (Kondrashov and Kondrashov 2001). An important disadvantage highlighted by the authors are linkage effects: without recombination to shuffle them up, beneficial mutations are linked to deleterious mutations in the same genome (hitchhiking) and purging deleterious mutations also purges neutral diversity across the genome (background selection). The authors propose the latter – efficient purifying selection and strong linkage – as an explanation for the low genetic diversity observed in M. tuberculosis. This is of course not exclusive of other related explanations, such as clonal interference (Gerrish and Lenski 1998). They also champion the use of forward evolutionary simulations (Haller and Messer 2019) to model the interplay between selection, recombination, and demography as a powerful alternative to traditional backward coalescent models. At times, Stritt and Gagneux are pessimistic about our existing methods – arguing that dN/dS and homoplasies “tell us little about the frequency and strength of selection.” Even though I favour a more optimistic view, I fully agree that our traditional population genetic metrics are sensitive to a slew of different deviations from a standard neutral evolution model and must be interpreted with caution. As I and others have argued, the extent of recombination (measured as the amount of linkage in a genome) is a key factor in determining how best to test for natural selection (Shapiro et al. 2009) and to conduct genotype-phenotype association studies (Chen and Shapiro 2021) in microbes. While this article is focused on the well-studied M. tuberculosis complex, there are many parallels with other clonal bacteria, including pathogens and symbionts. Whatever your favourite bug, we must all be careful to make sure the stories we tell about them are not “just so tales” but are supported, to the extent possible, by data and quantitative models. References Chen, Peter E., and B. Jesse Shapiro. 2021. "Classic Genome-Wide Association Methods Are Unlikely to Identify Causal Variants in Strongly Clonal Microbial Populations." bioRxiv. Stritt, C., Gagneux, S. (2023). How do monomorphic bacteria evolve? The Mycobacterium tuberculosis complex and the awkward population genetics of extreme clonality. EcoEvoRxiv, ver.3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.32942/X2GW2P | How do monomorphic bacteria evolve? The *Mycobacterium tuberculosis* complex and the awkward population genetics of extreme clonality | Christoph Stritt, Sebastien Gagneux | <p style="text-align: justify;">Exchange of genetic material through sexual reproduction or horizontal gene transfer is ubiquitous in nature. Among the few outliers that rarely recombine and mainly evolve by <em>de novo</em> mutation are a group o... | | Evolutionary Dynamics, Genome Evolution, Molecular Evolution, Population Genetics / Genomics, Reproduction and Sex | B. Jesse Shapiro | Gonçalo Themudo | 2022-12-16 13:41:53 | |
23 Jan 2020

A novel workflow to improve multi-locus genotyping of wildlife species: an experimental set-up with a known model systemImproving the reliability of genotyping of multigene families in non-model organismsRecommended by François Rousset based on reviews by Sebastian Ernesto Ramos-Onsins, Helena Westerdahl and Thomas BigotThe reliability of published scientific papers has been the topic of much recent discussion, notably in the biomedical sciences [1]. Although small sample size is regularly pointed as one of the culprits, big data can also be a concern. The advent of high-throughput sequencing, and the processing of sequence data by opaque bioinformatics workflows, mean that sequences with often high error rates are produced, and that exact but slow analyses are not feasible. References [1] Ioannidis, J. P. A, Greenland, S., Hlatky, M. A., Khoury, M. J., Macleod, M. R., Moher, D., Schulz, K. F. and Tibshirani, R. (2014) Increasing value and reducing waste in research design, conduct, and analysis. The Lancet, 383, 166-175. doi: 10.1016/S0140-6736(13)62227-8 | A novel workflow to improve multi-locus genotyping of wildlife species: an experimental set-up with a known model system | Gillingham, Mark A. F., Montero, B. Karina, Wilhelm, Kerstin, Grudzus, Kara, Sommer, Simone and Santos, Pablo S. C. | <p>Genotyping novel complex multigene systems is particularly challenging in non-model organisms. Target primers frequently amplify simultaneously multiple loci leading to high PCR and sequencing artefacts such as chimeras and allele amplification... | | Bioinformatics & Computational Biology, Evolutionary Ecology, Genome Evolution, Molecular Evolution | François Rousset | Helena Westerdahl, Sebastian Ernesto Ramos-Onsins, Paul J. McMurdie , Arnaud Estoup, Vincent Segura, Jacek Radwan , Torbjørn Rognes , William Stutz , Kevin Vanneste , Thomas Bigot, Jill A. Hollenbach , Wieslaw Babik , Marie-Christin... | 2019-05-15 17:30:44 | |
29 Sep 2017

Parallel diversifications of Cremastosperma and Mosannona (Annonaceae), tropical rainforest trees tracking Neogene upheaval of the South American continentUnravelling the history of Neotropical plant diversificationRecommended by Hervé Sauquet based on reviews by Thomas Couvreur and Hervé SauquetSouth American rainforests, particularly the Tropical Andes, have been recognized as the hottest spot of plant biodiversity on Earth, while facing unprecedented threats from human impact [1,2]. Considerable research efforts have recently focused on unravelling the complex geological, bioclimatic, and biogeographic history of the region [3,4]. While many studies have addressed the question of Neotropical plant diversification using parametric methods to reconstruct ancestral areas and patterns of dispersal, Pirie et al. [5] take a distinct, complementary approach. Based on a new, near-complete molecular phylogeny of two Neotropical genera of the flowering plant family Annonaceae, the authors modelled the ecological niche of each species and reconstructed the history of niche differentiation across the region. The main conclusion is that, despite similar current distributions and close phylogenetic distance, the two genera experienced rather distinct processes of diversification, responding differently to the major geological events marking the history of the region in the last 20 million years (Andean uplift, drainage of Lake Pebas, and closure of the Panama Isthmus). As a researcher who has not personally worked on Neotropical biogeography, I found this paper captivating and especially enjoyed very much reading the Introduction, which sets out the questions very clearly. The strength of this paper is the near-complete diversity of species the authors were able to sample in each clade and the high-quality data compiled for the niche models. I would recommend this paper as a nice example of a phylogenetic study aimed at unravelling the detailed history of Neotropical plant diversification. While large, synthetic meta-analyses of many clades should continue to seek general patterns [4,6], careful studies restricted on smaller, but well controlled and sampled datasets such as this one are essential to really understand tropical plant diversification in all its complexity. References [1] Antonelli A, and Sanmartín I. 2011. Why are there so many plant species in the Neotropics? Taxon 60, 403–414. [2] Mittermeier RA, Robles-Gil P, Hoffmann M, Pilgrim JD, Brooks TB, Mittermeier CG, Lamoreux JL and Fonseca GAB. 2004. Hotspots revisited: Earths biologically richest and most endangered ecoregions. CEMEX, Mexico City, Mexico 390pp [3] Antonelli A, Nylander JAA, Persson C and Sanmartín I. 2009. Tracing the impact of the Andean uplift on Neotropical plant evolution. Proceedings of the National Academy of Science of the USA 106, 9749–9754. doi: 10.1073/pnas.0811421106 [4] Hoorn C, Wesselingh FP, ter Steege H, Bermudez MA, Mora A, Sevink J, Sanmartín I, Sanchez-Meseguer A, Anderson CL, Figueiredo JP, Jaramillo C, Riff D, Negri FR, Hooghiemstra H, Lundberg J, Stadler T, Särkinen T and Antonelli A. 2010. Amazonia through time: Andean uplift, climate change, landscape evolution, and biodiversity. Science 330, 927–931. doi: 10.1126/science.1194585 [5] Pirie MD, Maas PJM, Wilschut R, Melchers-Sharrott H and Chatrou L. 2017. Parallel diversifications of Cremastosperma and Mosannona (Annonaceae), tropical rainforest trees tracking Neogene upheaval of the South American continent. bioRxiv, 141127, ver. 3 of 28th Sept 2017. doi: 10.1101/141127 [6] Bacon CD, Silvestro D, Jaramillo C, Tilston Smith B, Chakrabartye P and Antonelli A. 2015. Biological evidence supports an early and complex emergence of the Isthmus of Panama. Proceedings of the National Academy of Science of the USA 112, 6110–6115. doi: 10.1073/pnas.1423853112 | Parallel diversifications of Cremastosperma and Mosannona (Annonaceae), tropical rainforest trees tracking Neogene upheaval of the South American continent | Michael D. Pirie, Paul J. M. Maas, Rutger A. Wilschut, Heleen Melchers-Sharrott & Lars W. Chatrou | Much of the immense present day biological diversity of Neotropical rainforests originated from the Miocene onwards, a period of geological and ecological upheaval in South America. We assess the impact of the Andean orogeny, drainage of lake Peba... | | Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Hervé Sauquet | Hervé Sauquet, Thomas Couvreur | 2017-06-03 21:25:48 | |
15 Feb 2019

Architectural traits constrain the evolution of unisexual flowers and sexual segregation within inflorescences: an interspecific approachSometimes, sex is in the headRecommended by Juan Arroyo based on reviews by 3 anonymous reviewers based on reviews by 3 anonymous reviewers
Plants display an amazing diversity of reproductive strategies with and without sex. This diversity is particularly remarkable in flowering plants, as highlighted by Charles Darwin, who wrote several botanical books scrutinizing plant reproduction. One particularly influential work concerned floral variation [1]. Darwin recognized that flowers may present different forms within a single population, with or without sex specialization. The number of species concerned is small, but they display recurrent patterns, which made it possible for Darwin to invoke natural and sexual selection to explain them. Most of early evolutionary theory on the evolution of reproductive strategies was developed in the first half of the 20th century and was based on animals. However, the pioneering work by David Lloyd from the 1970s onwards excited interest in the diversity of plant sexual strategies as models for testing adaptive hypotheses and predicting reproductive outcomes [2]. The sex specialization of individual flowers and plants has since become one of the favorite topics of evolutionary biologists. However, attention has focused mostly on cases related to sex differentiation (dioecy and associated conditions [3]). Separate unisexual flower types on the same plant (monoecy and related cases, rendering the plant functionally hermaphroditic) have been much less studied, apart from their possible role in the evolution of dioecy [4] or their association with particular modes of pollination [5]. References [1] Darwin, C. (1877). The different forms of flowers on plants of the same species. John Murray. | Architectural traits constrain the evolution of unisexual flowers and sexual segregation within inflorescences: an interspecific approach | Rubén Torices, Ana Afonso, Arne A. Anderberg, José M. Gómez and Marcos Méndez | <p>Male and female unisexual flowers have repeatedly evolved from the ancestral bisexual flowers in different lineages of flowering plants. This sex specialization in different flowers often occurs within inflorescences. We hypothesize that inflor... | | Evolutionary Ecology, Morphological Evolution, Phenotypic Plasticity, Reproduction and Sex, Sexual Selection | Juan Arroyo | Jana Vamosi, Marcial Escudero, Anonymous | 2018-06-27 10:49:52 | |
05 Apr 2024
Does the seed fall far from the tree? Weak fine scale genetic structure in a continuous Scots pine populationWeak spatial genetic structure in a large continuous Scots pine population – implications for conservation and breedingRecommended by Myriam Heuertz based on reviews by Joachim Mergeay, Jean-Baptiste Ledoux and Roberta Loh
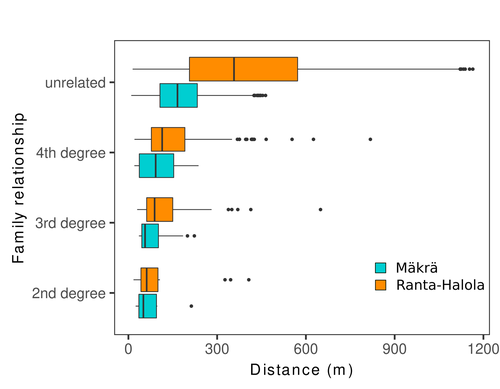
Spatial genetic structure, i.e. the non-random spatial distribution of genotypes, arises in populations because of different processes including spatially limited dispersal and selection. Knowledge on the spatial genetic structure of plant populations is important to assess biological parameters such as gene dispersal distances and the potential for local adaptations, as well as for applications in conservation management and breeding. In their work, Niskanen and colleagues demonstrate a multifaceted approach to characterise the spatial genetic structure in two replicate sites of a continuously distributed Scots pine population in South-Eastern Finland. They mapped and assessed the ages of 469 naturally regenerated adults and genotyped them using a SNP array which resulted in 157 325 filtered polymorphic SNPs. Their dataset is remarkably powerful because of the large numbers of both individuals and SNPs genotyped. This made it possible to characterise precisely the decay of genetic relatedness between individuals with spatial distance despite the extensive dispersal capacity of Scots pine through pollen, and ensuing expectations of an almost panmictic population. The authors’ data analysis was particularly thorough. They demonstrated that two metrics of pairwise relatedness, the genomic relationship matrix (GRM, Yang et al. 2011) and the kinship coefficient (Loiselle et al. 1995) were strongly correlated and produced very similar inference of family relationships: >99% of pairs of individuals were unrelated, and the remainder exhibited 2nd (e.g., half-siblings) to 4th degree relatedness. Pairwise relatedness decayed with spatial distance which resulted in extremely weak but statistically significant spatial genetic structure in both sites, quantified as Sp=0.0005 and Sp=0.0008. These estimates are at least an order of magnitude lower than estimates in the literature obtained in more fragmented populations of the same species or in other conifers. Estimates of the neighbourhood size, the effective number of potentially mating individuals belonging to a within-population neighbourhood (Wright 1946), were relatively large with Nb=1680-3210 despite relatively short gene dispersal distances, σg = 36.5–71.3m, which illustrates the high effective density of the population. The authors showed the implications of their findings for selection. The capacity for local adaptation depends on dispersal distances and the strength of the selection coefficient. In the study population, the authors inferred that local adaptation can only occur if environmental heterogeneity occurs over a distance larger than approximately one kilometre (or larger, if considering long-distance dispersal). Interestingly, in Scots pine, no local adaptation has been described on similar geographic scales, in contrast to some other European or Mediterranean conifers (Scotti et al. 2023). The authors’ results are relevant for the management of conservation and breeding. They showed that related individuals occurred within sites only and that they shared a higher number of rare alleles than unrelated ones. Since rare alleles are enriched in new and recessive deleterious variants, selecting related individuals could have negative consequences in breeding programmes. The authors also showed, in their response to reviewers, that their powerful dataset was not suitable to obtain a robust estimate of effective population size, Ne, based on the linkage disequilibrium method (Do et al. 2014). This illustrated that the estimation of Ne used for genetic indicators supported in international conservation policy (Hoban et al. 2020, CBD 2022) remains challenging in large and continuous populations (see also Santo-del-Blanco et al. 2023, Gargiulo et al. 2024). ReferencesCBD (2022) Kunming-Montreal Global Biodiversity Framework. https://www.cbd.int/doc/decisions/cop-15/cop-15-dec-04-en.pdf Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014). NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne ) from genetic data. Molecular Ecology Resources 14: 209–214. https://doi.org/10.1111/1755-0998.12157 Gargiulo R, Decroocq V, González-Martínez SC, Paz-Vinas I, Aury JM, Kupin IL, Plomion C, Schmitt S, Scotti I, Heuertz M (2024) Estimation of contemporary effective population size in plant populations: limitations of genomic datasets. Evolutionary Applications, in press, https://doi.org/10.1101/2023.07.18.549323 Hoban S, Bruford M, D’Urban Jackson J, Lopes-Fernandes M, Heuertz M, Hohenlohe PA, Paz-Vinas I, et al. (2020) Genetic diversity targets and indicators in the CBD post-2020 Global Biodiversity Framework must be improved. Biological Conservation 248: 108654. https://doi.org/10.1016/j.biocon.2020.108654 Loiselle BA, Sork VL, Nason J & Graham C (1995) Spatial genetic structure of a tropical understorey shrub, Psychotria officinalis (Rubiaceae). American Journal of Botany 82: 1420–1425. https://doi.org/10.1002/j.1537-2197.1995.tb12679.x Santos-del-Blanco L, Olsson S, Budde KB, Grivet D, González-Martínez SC, Alía R, Robledo-Arnuncio JJ (2022). On the feasibility of estimating contemporary effective population size (Ne) for genetic conservation and monitoring of forest trees. Biological Conservation 273: 109704. https://doi.org/10.1016/j.biocon.2022.109704 Scotti I, Lalagüe H, Oddou-Muratorio S, Scotti-Saintagne C, Ruiz Daniels R, Grivet D, et al. (2023) Common microgeographical selection patterns revealed in four European conifers. Molecular Ecology 32: 393-411. https://doi.org/10.1111/mec.16750 Wright S (1946) Isolation by distance under diverse systems of mating. Genetics 31: 39–59. https://doi.org/10.1093/genetics/31.1.39 Yang J, Lee SH, Goddard ME & Visscher PM (2011) GCTA: a tool for genome-wide complex trait analysis. The American Journal of Human Genetics 88: 76–82. https://www.cell.com/ajhg/pdf/S0002-9297(10)00598-7.pdf | Does the seed fall far from the tree? Weak fine scale genetic structure in a continuous Scots pine population | Alina K. Niskanen, Sonja T. Kujala, Katri Kärkkäinen, Outi Savolainen, Tanja Pyhäjärvi | <p>Knowledge of fine-scale spatial genetic structure, i.e., the distribution of genetic diversity at short distances, is important in evolutionary research and in practical applications such as conservation and breeding programs. In trees, related... | | Adaptation, Evolutionary Applications, Population Genetics / Genomics | Myriam Heuertz | Joachim Mergeay | 2023-06-27 21:57:28 | |
07 Sep 2018

Parallel pattern of differentiation at a genomic island shared between clinal and mosaic hybrid zones in a complex of cryptic seahorse lineagesGenomic parallelism in adaptation to orthogonal environments in sea horsesRecommended by Yaniv Brandvain based on reviews by 2 anonymous reviewersStudies in speciation genomics have revealed that gene flow is quite common, and that despite this, species can maintain their distinct environmental adaptations. Although researchers are still elucidating the genomic mechanisms by which species maintain their adaptations in the face of gene flow, this often appears to involve few diverged genomic regions in otherwise largely undifferentiated genomes. In this preprint [1], Riquet and colleagues investigate the genetic structuring and patterns of parallel evolution in the long-snouted seahorse. References [1] Riquet, F., Liautard-Haag, C., Woodall, L., Bouza, C., Louisy, P., Hamer, B., Otero-Ferrer, F., Aublanc, P., Béduneau, V., Briard, O., El Ayari, T., Hochscheid, S. Belkhir, K., Arnaud-Haond, S., Gagnaire, P.-A., Bierne, N. (2018). Parallel pattern of differentiation at a genomic island shared between clinal and mosaic hybrid zones in a complex of cryptic seahorse lineages. bioRxiv, 161786, ver. 4 recommended and peer-reviewed by PCI Evol Biol. doi: 10.1101/161786 | Parallel pattern of differentiation at a genomic island shared between clinal and mosaic hybrid zones in a complex of cryptic seahorse lineages | Florentine Riquet, Cathy Liautard-Haag, Lucy Woodall, Carmen Bouza, Patrick Louisy, Bojan Hamer, Francisco Otero-Ferrer, Philippe Aublanc, Vickie Béduneau, Olivier Briard, Tahani El Ayari, Sandra Hochscheid, Khalid Belkhir, Sophie Arnaud-Haond, Pi... | <p>Diverging semi-isolated lineages either meet in narrow clinal hybrid zones, or have a mosaic distribution associated with environmental variation. Intrinsic reproductive isolation is often emphasized in the former and local adaptation in the la... | | Hybridization / Introgression, Molecular Evolution, Population Genetics / Genomics, Speciation | Yaniv Brandvain | Kathleen Lotterhos, Sarah Fitzpatrick | 2017-07-11 13:12:40 | |
16 Mar 2023
Phylogeographic breaks and how to find them: Separating vicariance from isolation by distance in a lizard with restricted dispersalThe difficult task of partitioning the effects of vicariance and isolation by distance in poor dispersersRecommended by Eric Pante based on reviews by Kevin Sánchez and Aglaia (Cilia) Antoniou
Partitioning the effects of vicariance and low dispersal has been a long-standing problem in historical biogeography and phylogeography. While the term “vicariance” refers to divergence in allopatry, caused by some physical (geological, geographical) or climatic barriers (e.g. Rosen 1978), isolation by distance refers to the genetic differentiation of remote populations due to the physical distance separating them, when the latter surpasses the scale of dispersal (Wright 1938, 1940, 1943). Vicariance and dispersal have long been considered as separate forces leading to separate scenarii of speciation (e.g. reviewed in Hickerson and Meyer 2008). Nevertheless, these two processes are strongly linked, as, for example, vicariance theory relies on the assumption that ancestral lineages were once linked by dispersal prior to physical or climatic isolation (Rosen 1978). Low dispersal and vicariance are not mutually exclusive, and distinguishing these two processes in heterogeneous landscapes, especially for poor dispersers, remains therefore a severe challenge. For example, low dispersal (and/or small population size) can give rise to geographic patterns consistent with a phylogeographic break and be mistaken for geographic isolation (Irwin 2002, Kuo and Avise 2005). The study of Rancilliac and colleagues (2023) is at the heart of this issue. It focuses on a nominal lizard species, the red-tailed spiny-footed lizard (Acanthodactylus erythrurus, Squamata: Lacertidae), which has a wide spatial distribution (from the Maghreb to the Iberian Peninsula), is found in a variety of different habitats, and has a wide range of morphological traits that do not always correlate with phylogeny. The main question is the following: have “the morphological and ecological diversification of this group been produced by vicariance and lineage diversification, or by local adaptation in the face of historical gene flow?” To tackle this question, the authors used sequence data from multiple mitochondrial and nuclear markers and a nested analysis workflow integrating phylogeography, multiple correspondence analyses and a relatively novel approach to IBD testing (Hausdorf & Henning, 2020). The latter is based on regression analysis and was shown to be less prone to error than the traditional (partial) Mantel test. While this set of methods allowed the partitioning of the effect of isolation by distance and vicariance in shaping contemporary genetic diversity in red-tailed spiny-footed lizards, some of the evolutionary history of this species complex remains blurred by ongoing gene flow and admixture, retention of ancestral polymorphism, or selection. The lack of congruence between mitochondrial and nuclear gene trees once again warns us that proposing evolutionary scenarii based on individual gene trees is a risky business. References Hausdorf B, Hennig C (2020) Species delimitation and geography. Molecular Ecology Resources, 20, 950–960. https://doi.org/10.1111/1755-0998.13184 Hickerson MJ, Meyer CP (2008) Testing comparative phylogeographic models of marine vicariance and dispersal using a hierarchical Bayesian approach. BMC Evolutionary Biology, 8, 322. https://doi.org/10.1186/1471-2148-8-322 Irwin DE (2002) Phylogeographic breaks without geographic barriers to gene flow. Evolution, 56, 2383–2394. https://doi.org/10.1111/j.0014-3820.2002.tb00164.x Kuo C-H, Avise JC (2005) Phylogeographic breaks in low-dispersal species: the emergence of concordance across gene trees. Genetica, 124, 179–186. https://doi.org/10.1007/s10709-005-2095-y Rancilhac L, Miralles A, Geniez P, Mendez-Aranda D, Beddek M, Brito JC, Leblois R, Crochet P-A (2023) Phylogeographic breaks and how to find them: An empirical attempt at separating vicariance from isolation by distance in a lizard with restricted dispersal. bioRxiv, 2022.09.30.510256, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.09.30.510256 Rosen DE (1978) Vicariant Patterns and Historical Explanation in Biogeography. Systematic Biology, 27, 159–188. https://doi.org/10.2307/2412970 Wright, S (1938) Size of population and breeding structure in relation to evolution. Science 87:430-431. Wright S (1940) Breeding Structure of Populations in Relation to Speciation. The American Naturalist, 74, 232–248. https://doi.org/10.1086/280891 Wright S (1943) Isolation by distance. Genetics, 28, 114–138. https://doi.org/10.1093/genetics/28.2.114 | Phylogeographic breaks and how to find them: Separating vicariance from isolation by distance in a lizard with restricted dispersal | Loïs Rancilhac, Aurélien Miralles, Philippe Geniez, Daniel Mendez-Arranda, Menad Beddek, José Carlos Brito, Raphaël Leblois, Pierre-André Crochet | <p>Aim</p> <p>Discontinuity in the distribution of genetic diversity (often based on mtDNA) is usually interpreted as evidence for phylogeographic breaks, underlying vicariant units. However, a misleading signal of phylogeographic break can arise... | | Phylogeography & Biogeography, Population Genetics / Genomics, Speciation, Systematics / Taxonomy | Eric Pante | Kevin Sánchez | 2022-10-05 13:11:28 | |
06 Oct 2017

Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebratesCombining molecular information on chromatin organisation with eQTLs and evolutionary conservation provides strong candidates for the evolution of gene regulation in mammalian brainsRecommended by Marc Robinson-Rechavi based on reviews by Marc Robinson-Rechavi and Charles DankoIn this manuscript [1], Francisco J. Novo proposes candidate non-coding genomic elements regulating neurodevelopmental genes. What is very nice about this study is the way in which public molecular data, including physical interaction data, is used to leverage recent advances in our understanding to molecular mechanisms of gene regulation in an evolutionary context. More specifically, evolutionarily conserved non coding sequences are combined with enhancers from the FANTOM5 project, DNAse hypersensitive sites, chromatin segmentation, ChIP-seq of transcription factors and of p300, gene expression and eQTLs from GTEx, and physical interactions from several Hi-C datasets. The candidate regulatory regions thus identified are linked to candidate regulated genes, and the author shows their potential implication in brain development. While the results are focused on a small number of genes, this allows to verify features of these candidates in great detail. This study shows how functional genomics is increasingly allowing us to fulfill the promises of Evo-Devo: understanding the molecular mechanisms of conservation and differences in morphology. References [1] Novo, FJ. 2017. Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebrates. bioRxiv, 150482, ver. 4 of Sept 29th, 2017. doi: 10.1101/150482 | Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebrates | Francisco J. Novo | <p>Many non-coding regulatory elements conserved in vertebrates regulate the expression of genes involved in development and play an important role in the evolution of morphology through the rewiring of developmental gene networks. Available biolo... | | Genome Evolution | Marc Robinson-Rechavi | Marc Robinson-Rechavi, Charles Danko | 2017-06-29 08:55:41 | |
09 Dec 2019

Systematics and geographical distribution of Galba species, a group of cryptic and worldwide freshwater snailsThe challenge of delineating species when they are hiddenRecommended by Fabien Condamine based on reviews by Pavel Matos, Christelle Fraïsse and Niklas WahlbergThe science of naming species (taxonomy) has been renewed with the developments of molecular sequencing, digitization of museum specimens, and novel analytical tools. However, naming species can be highly subjective, sometimes considered as an art [1], because it is based on human-based criteria that vary among taxonomists. Nonetheless, taxonomists often argue that species names are hypotheses, which are therefore testable and refutable as new evidence is provided. This challenge comes with a more and more recognized and critical need for rigorously delineated species not only for producing accurate species inventories, but more importantly many questions in evolutionary biology (e.g. speciation), ecology (e.g. ecosystem structure and functioning), conservation biology (e.g. targeting priorities) or biogeography (e.g. diversification processes) depend in part on those species inventories and our knowledge of species [2-3]. Inaccurate species boundaries or diversity estimates may lead us to deliver biased answers to those questions, exactly as phylogenetic trees must be reconstructed rigorously and analyzed critically because they are a first step toward discussing broader questions [2-3]. In this context, biological diversity needs to be studied from multiple and complementary perspectives requiring the collaboration of morphologists, molecular biologists, biogeographers, and modelers [4-5]. Integrative taxonomy has been proposed as a solution to tackle the challenge of delimiting species [2], especially in highly diverse and undocumented groups of organisms. References [1] Ohl, M. (2018). The art of naming. MIT Press. | Systematics and geographical distribution of Galba species, a group of cryptic and worldwide freshwater snails | Pilar Alda, Manon Lounnas, Antonio Alejandro Vázquez, Rolando Ayaqui, Manuel Calvopina, Maritza Celi-Erazo, Robert Dillon, Luisa Carolina González Ramírez, Eric S. Loker, Jenny Muzzio-Aroca, Alberto Orlando Nárvaez, Oscar Noya, Andrés Esteban Pere... | <p>Cryptic species can present a significant challenge to the application of systematic and biogeographic principles, especially if they are invasive or transmit parasites or pathogens. Detecting cryptic species requires a pluralistic approach in ... | | Phylogeography & Biogeography, Systematics / Taxonomy | Fabien Condamine | Pavel Matos, Christelle Fraïsse | 2019-05-25 10:34:57 |




