
Latest recommendations

| Id | Title▲ | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
11 Jul 2022

Mutualists construct the ecological conditions that trigger the transition from parasitismGive them some space: how spatial structure affects the evolutionary transition towards mutualistic symbiosisRecommended by Francois Massol based on reviews by Eva Kisdi and 3 anonymous reviewers based on reviews by Eva Kisdi and 3 anonymous reviewers
The evolution of mutualistic symbiosis is a puzzle that has fascinated evolutionary ecologist for quite a while. Data on transitions between symbiotic bacterial ways of life has evidenced shifts from mutualism towards parasitism and vice versa (Sachs et al., 2011), so there does not seem to be a strong determinism on those transitions. From the host’s perspective, mutualistic symbiosis implies at the very least some form of immune tolerance, which can be costly (e.g. Sorci, 2013). Empirical approaches thus raise very important questions: How can symbiosis turn from parasitism into mutualism when it seemingly needs such a strong alignment of selective pressures on both the host and the symbiont? And yet why is mutualistic symbiosis so widespread and so important to the evolution of macro-organisms (Margulis, 1998)? While much of the theoretical literature on the evolution of symbiosis and mutualism has focused on either the stability of such relationships when non-mutualists can invade the host-symbiont system (e.g. Ferrière et al., 2007) or the effect of the mode of symbiont transmission on the evolutionary dynamics of mutualism (e.g. Genkai-Kato and Yamamura, 1999), the question remains whether and under which conditions parasitic symbiosis can turn into mutualism in the first place. Earlier results suggested that spatial demographic heterogeneity between host populations could be the leading determinant of evolution towards mutualism or parasitism (Hochberg et al., 2000). Here, Ledru et al. (2022) investigate this question in an innovative way by simulating host-symbiont evolutionary dynamics in a spatially explicit context. Their hypothesis is intuitive but its plausibility is difficult to gauge without a model: Does the evolution towards mutualism depend on the ability of the host and symbiont to evolve towards close-range dispersal in order to maintain clusters of efficient host-symbiont associations, thus outcompeting non-mutualists? I strongly recommend reading this paper as the results obtained by the authors are very clear: competition strength and the cost of dispersal both affect the likelihood of the transition from parasitism to mutualism, and once mutualism has set in, symbiont trait values clearly segregate between highly dispersive parasites and philopatric mutualists. The demonstration of the plausibility of their hypothesis is accomplished with brio and thoroughness as the authors also examine the conditions under which the transition can be reversed, the impact of the spatial range of competition and the effect of mortality. Since high dispersal cost and strong, long-range competition appear to be the main factors driving the evolutionary transition towards mutualistic symbiosis, now is the time for empiricists to start investigating this question with spatial structure in mind. References Ferrière, R., Gauduchon, M. and Bronstein, J. L. (2007) Evolution and persistence of obligate mutualists and exploiters: competition for partners and evolutionary immunization. Ecology Letters, 10, 115-126. https://doi.org/10.1111/j.1461-0248.2006.01008.x Genkai-Kato, M. and Yamamura, N. (1999) Evolution of mutualistic symbiosis without vertical transmission. Theoretical Population Biology, 55, 309-323. https://doi.org/10.1006/tpbi.1998.1407 Hochberg, M. E., Gomulkiewicz, R., Holt, R. D. and Thompson, J. N. (2000) Weak sinks could cradle mutualistic symbioses - strong sources should harbour parasitic symbioses. Journal of Evolutionary Biology, 13, 213-222. https://doi.org/10.1046/j.1420-9101.2000.00157.x Ledru L, Garnier J, Rohr M, Noûs C and Ibanez S (2022) Mutualists construct the ecological conditions that trigger the transition from parasitism. bioRxiv, 2021.08.18.456759, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.08.18.456759 Margulis, L. (1998) Symbiotic planet: a new look at evolution, Basic Books, Amherst. Sachs, J. L., Skophammer, R. G. and Regus, J. U. (2011) Evolutionary transitions in bacterial symbiosis. Proceedings of the National Academy of Sciences, 108, 10800-10807. https://doi.org/10.1073/pnas.1100304108 Sorci, G. (2013) Immunity, resistance and tolerance in bird–parasite interactions. Parasite Immunology, 35, 350-361. https://doi.org/10.1111/pim.12047 | Mutualists construct the ecological conditions that trigger the transition from parasitism | Leo Ledru, Jimmy Garnier, Matthias Rohr, Camille Nous, Sebastien Ibanez | <p>The evolution of mutualism between hosts and initially parasitic symbionts represents a major transition in evolution. Although vertical transmission of symbionts during host reproduction and partner control both favour the stability of mutuali... | | Evolutionary Ecology, Species interactions | Francois Massol | 2021-08-20 12:25:40 | ||
19 Mar 2018

Natural selection on plasticity of thermal traits in a highly seasonal environmentIs thermal plasticity itself shaped by natural selection? An assessment with desert frogsRecommended by Wolf Blanckenhorn based on reviews by Dries Bonte, Wolf Blanckenhorn and Nadia Aubin-HorthIt is well known that climatic factors – most notably temperature, season length, insolation and humidity – shape the thermal niche of organisms on earth through the action of natural selection. But how is this achieved precisely? Much of thermal tolerance is actually mediated by phenotypic plasticity (as opposed to genetic adaptation). A prominent expectation is that environments with greater (daily and/or annual) thermal variability select for greater plasticity, i.e. better acclimation capacity. Thus, plasticity might be selected per se. A Chilean group around Leonardo Bacigalupe assessed natural selection in the wild in one marginal (and extreme) population of the four-eyed frog Pleurodema thaul (Anura: Leptodactylidae) in an isolated oasis in the Atacama Desert, permitting estimation of mortality without much potential of confounding it with migration [1]. Several thermal traits were considered: CTmax – the critical maximal temperature; CTmin – the critical minimum temperature; Tpref – preferred temperature; Q10 – thermal sensitivity of metabolism; and body mass. Animals were captured in the wild and subsequently assessed for thermal traits in the laboratory at two acclimation temperatures (10° & 20°C), defining the plasticity in all traits as the difference between the traits at the two acclimation temperatures. Thereafter the animals were released again in their natural habitat and their survival was monitored over the subsequent 1.5 years, covering two breeding seasons, to estimate viability selection in the wild. The authors found and conclude that, aside from larger body size increasing survival (an unsurprising result), plasticity does not seem to be systematically selected directly, while some of the individual traits show weak signs of selection. Despite limited sample size (ca. 80 frogs) investigated in only one marginal but very seasonal population, this study is interesting because selection on plasticity in physiological thermal traits, as opposed to selection on the thermal traits themselves, is rarely investigated. The study thus also addressed the old but important question of whether plasticity (i.e. CTmax-CTmin) is a trait by itself or an epiphenomenon defined by the actual traits (CTmax and CTmin) [2-5]. Given negative results, the main question could not be ultimately solved here, so more similar studies should be performed. References [1] Bacigalupe LD, Gaitan-Espitia, JD, Barria AM, Gonzalez-Mendez A, Ruiz-Aravena M, Trinder M & Sinervo B. 2018. Natural selection on plasticity of thermal traits in a highly seasonal environment. bioRxiv 191825, ver. 5 peer-reviewed by Peer Community In Evolutionary Biology. doi: 10.1101/191825 | Natural selection on plasticity of thermal traits in a highly seasonal environment | Leonardo Bacigalupe, Juan Diego Gaitan-Espitia, Aura M Barria, Avia Gonzalez-Mendez, Manuel Ruiz-Aravena, Mark Trinder, Barry Sinervo | <p>For ectothermic species with broad geographical distributions, latitudinal/altitudinal variation in environmental temperatures (averages and extremes) are expected to shape the evolution of physiological tolerances and the acclimation capacity ... | | Adaptation, Evolutionary Ecology, Phenotypic Plasticity | Wolf Blanckenhorn | 2017-09-22 23:17:40 | ||
07 Jul 2017

Negative frequency-dependent selection is frequently confoundingUnmasking the delusive appearance of negative frequency-dependent selectionRecommended by Ignacio Bravo based on reviews by David Baltrus and 2 anonymous reviewersExplaining the processes that maintain polymorphisms in a population has been a fundamental line of research in evolutionary biology. One of the main mechanisms identified that preserves genetic diversity is negative frequency-dependent selection (NFDS), which constitutes a powerful framework for interpreting the presence of persistent polymorphisms. Nevertheless, a number of patterns that are often explained by invoking NFDS may also be compatible with, and possibly more easily explained by, different processes. References [1] Brisson D. 2017. Negative frequency-dependent selection is frequently confounding. bioRxiv 113324, ver. 3 of 20th June 2017. doi: 10.1101/113324 [2] Heino M, Metz JAJ and Kaitala V. 1998. The enigma of frequency-dependent selection. Trends in Ecology & Evolution 13: 367-370. doi: 1016/S0169-5347(98)01380-9 | Negative frequency-dependent selection is frequently confounding | Dustin Brisson | The existence of persistent genetic variation within natural populations presents an evolutionary problem as natural selection and genetic drift tend to erode genetic diversity. Models of balancing selection were developed to account for the high ... | | Evolutionary Applications, Evolutionary Theory, Population Genetics / Genomics | Ignacio Bravo | 2017-03-03 18:46:42 | ||
07 Nov 2019

New insights into the population genetics of partially clonal organisms: when seagrass data meet theoretical expectationsInferring rates of clonal versus sexual reproduction from population genetics dataRecommended by Olivier J Hardy based on reviews by Ludwig TRIEST, Stacy Krueger-Hadfield and 1 anonymous reviewerIn partially clonal organisms, genetic markers are often used to characterize the genotypic diversity of populations and infer thereof the relative importance of clonal versus sexual reproduction. Most studies report a measure of genotypic diversity based on a ratio, R, of the number of distinct multilocus genotypes over the sample size, and qualitatively interpret high / low R as indicating the prevalence of sexual / clonal reproduction. However, a theoretical framework allowing to quantify the relative rates of clonal versus sexual reproduction from genotypic diversity is still lacking, except using temporal sampling. Moreover, R is intrinsically highly dependent on sample size and sample design, while alternative measures of genotypic diversity are more robust to sample size, like D*, which is equivalent to the Gini-Simpson diversity index applied to multilocus genotypes. Another potential indicator of reproductive strategies is the inbreeding coefficient, Fis, because population genetics theory predicts that clonal reproduction should lead to negative Fis, at least when the sexual reproduction component occurs through random mating. Taking advantage of this prediction, Arnaud-Haond et al. [1] reanalysed genetic data from 165 populations of four partially clonal seagrass species sampled in a standardized way. They found positive correlations between Fis and both R and D* within each species, reflecting variation in the relative rates of sexual versus clonal reproduction among populations. Moreover, the differences of mean genotypic diversity and Fis values among species were also consistent with their known differences in reproductive strategies. Arnaud-Haond et al. [1] also conclude that previous works based on the interpretation of R generally lead to underestimate the prevalence of clonality in seagrasses. Arnaud-Haond et al. [1] confirm experimentally that Fis merits to be interpreted more properly than usually done when inferring rates of clonal reproduction from population genetics data of species reproducing both sexually and clonally. An advantage of Fis is that it is much less affected by sample size than R, and thus should be more reliable when comparing studies differing in sample design. Hence, when the rate of clonal reproduction becomes significant, we expect Fis < 0 and D* < 1. I expect these two indicators of clonality to be complementary because they rely on different consequences of clonality on pattern of genetic variation. Nevertheless, both measures can be affected by other factors. For example, null alleles, selfing or biparental inbreeding can pull Fis upwards, potentially eliminating the signature of clonal reproduction. Similarly, D* (and other measures of genotypic diversity) can be low because the polymorphism of the genetic markers used is too limited or because sexual reproduction often occurs through selfing, eventually resulting in highly similar homozygous genotypes. References [1] Arnaud-Haond, S., Stoeckel, S., and Bailleul, D. (2019). New insights into the population genetics of partially clonal organisms: when seagrass data meet theoretical expectations. ArXiv:1902.10240 [q-Bio], v6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. Retrieved from http://arxiv.org/abs/1902.10240 | New insights into the population genetics of partially clonal organisms: when seagrass data meet theoretical expectations | Arnaud-Haond, Sophie, Stoeckel, Solenn, and Bailleul, Diane | <p>Seagrass meadows are among the most important coastal ecosystems, in terms of both spatial extent and ecosystem services, but they are also declining worldwide. Understanding the drivers of seagrass meadow dynamics is essential for designing so... | | Evolutionary Ecology, Population Genetics / Genomics, Reproduction and Sex | Olivier J Hardy | 2019-03-01 21:57:34 | ||
29 Sep 2017

Parallel diversifications of Cremastosperma and Mosannona (Annonaceae), tropical rainforest trees tracking Neogene upheaval of the South American continentUnravelling the history of Neotropical plant diversificationRecommended by Hervé Sauquet based on reviews by Thomas Couvreur and Hervé SauquetSouth American rainforests, particularly the Tropical Andes, have been recognized as the hottest spot of plant biodiversity on Earth, while facing unprecedented threats from human impact [1,2]. Considerable research efforts have recently focused on unravelling the complex geological, bioclimatic, and biogeographic history of the region [3,4]. While many studies have addressed the question of Neotropical plant diversification using parametric methods to reconstruct ancestral areas and patterns of dispersal, Pirie et al. [5] take a distinct, complementary approach. Based on a new, near-complete molecular phylogeny of two Neotropical genera of the flowering plant family Annonaceae, the authors modelled the ecological niche of each species and reconstructed the history of niche differentiation across the region. The main conclusion is that, despite similar current distributions and close phylogenetic distance, the two genera experienced rather distinct processes of diversification, responding differently to the major geological events marking the history of the region in the last 20 million years (Andean uplift, drainage of Lake Pebas, and closure of the Panama Isthmus). As a researcher who has not personally worked on Neotropical biogeography, I found this paper captivating and especially enjoyed very much reading the Introduction, which sets out the questions very clearly. The strength of this paper is the near-complete diversity of species the authors were able to sample in each clade and the high-quality data compiled for the niche models. I would recommend this paper as a nice example of a phylogenetic study aimed at unravelling the detailed history of Neotropical plant diversification. While large, synthetic meta-analyses of many clades should continue to seek general patterns [4,6], careful studies restricted on smaller, but well controlled and sampled datasets such as this one are essential to really understand tropical plant diversification in all its complexity. References [1] Antonelli A, and Sanmartín I. 2011. Why are there so many plant species in the Neotropics? Taxon 60, 403–414. [2] Mittermeier RA, Robles-Gil P, Hoffmann M, Pilgrim JD, Brooks TB, Mittermeier CG, Lamoreux JL and Fonseca GAB. 2004. Hotspots revisited: Earths biologically richest and most endangered ecoregions. CEMEX, Mexico City, Mexico 390pp [3] Antonelli A, Nylander JAA, Persson C and Sanmartín I. 2009. Tracing the impact of the Andean uplift on Neotropical plant evolution. Proceedings of the National Academy of Science of the USA 106, 9749–9754. doi: 10.1073/pnas.0811421106 [4] Hoorn C, Wesselingh FP, ter Steege H, Bermudez MA, Mora A, Sevink J, Sanmartín I, Sanchez-Meseguer A, Anderson CL, Figueiredo JP, Jaramillo C, Riff D, Negri FR, Hooghiemstra H, Lundberg J, Stadler T, Särkinen T and Antonelli A. 2010. Amazonia through time: Andean uplift, climate change, landscape evolution, and biodiversity. Science 330, 927–931. doi: 10.1126/science.1194585 [5] Pirie MD, Maas PJM, Wilschut R, Melchers-Sharrott H and Chatrou L. 2017. Parallel diversifications of Cremastosperma and Mosannona (Annonaceae), tropical rainforest trees tracking Neogene upheaval of the South American continent. bioRxiv, 141127, ver. 3 of 28th Sept 2017. doi: 10.1101/141127 [6] Bacon CD, Silvestro D, Jaramillo C, Tilston Smith B, Chakrabartye P and Antonelli A. 2015. Biological evidence supports an early and complex emergence of the Isthmus of Panama. Proceedings of the National Academy of Science of the USA 112, 6110–6115. doi: 10.1073/pnas.1423853112 | Parallel diversifications of Cremastosperma and Mosannona (Annonaceae), tropical rainforest trees tracking Neogene upheaval of the South American continent | Michael D. Pirie, Paul J. M. Maas, Rutger A. Wilschut, Heleen Melchers-Sharrott & Lars W. Chatrou | Much of the immense present day biological diversity of Neotropical rainforests originated from the Miocene onwards, a period of geological and ecological upheaval in South America. We assess the impact of the Andean orogeny, drainage of lake Peba... | | Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Hervé Sauquet | Hervé Sauquet, Thomas Couvreur | 2017-06-03 21:25:48 | |
07 Sep 2018

Parallel pattern of differentiation at a genomic island shared between clinal and mosaic hybrid zones in a complex of cryptic seahorse lineagesGenomic parallelism in adaptation to orthogonal environments in sea horsesRecommended by Yaniv Brandvain based on reviews by 2 anonymous reviewersStudies in speciation genomics have revealed that gene flow is quite common, and that despite this, species can maintain their distinct environmental adaptations. Although researchers are still elucidating the genomic mechanisms by which species maintain their adaptations in the face of gene flow, this often appears to involve few diverged genomic regions in otherwise largely undifferentiated genomes. In this preprint [1], Riquet and colleagues investigate the genetic structuring and patterns of parallel evolution in the long-snouted seahorse. References [1] Riquet, F., Liautard-Haag, C., Woodall, L., Bouza, C., Louisy, P., Hamer, B., Otero-Ferrer, F., Aublanc, P., Béduneau, V., Briard, O., El Ayari, T., Hochscheid, S. Belkhir, K., Arnaud-Haond, S., Gagnaire, P.-A., Bierne, N. (2018). Parallel pattern of differentiation at a genomic island shared between clinal and mosaic hybrid zones in a complex of cryptic seahorse lineages. bioRxiv, 161786, ver. 4 recommended and peer-reviewed by PCI Evol Biol. doi: 10.1101/161786 | Parallel pattern of differentiation at a genomic island shared between clinal and mosaic hybrid zones in a complex of cryptic seahorse lineages | Florentine Riquet, Cathy Liautard-Haag, Lucy Woodall, Carmen Bouza, Patrick Louisy, Bojan Hamer, Francisco Otero-Ferrer, Philippe Aublanc, Vickie Béduneau, Olivier Briard, Tahani El Ayari, Sandra Hochscheid, Khalid Belkhir, Sophie Arnaud-Haond, Pi... | <p>Diverging semi-isolated lineages either meet in narrow clinal hybrid zones, or have a mosaic distribution associated with environmental variation. Intrinsic reproductive isolation is often emphasized in the former and local adaptation in the la... | | Hybridization / Introgression, Molecular Evolution, Population Genetics / Genomics, Speciation | Yaniv Brandvain | Kathleen Lotterhos, Sarah Fitzpatrick | 2017-07-11 13:12:40 | |
14 Apr 2021

Parasitic success and venom composition evolve upon specialization of parasitoid wasps to different host speciesWhat makes a parasite successful? Parasitoid wasp venoms evolve rapidly in a host-specific mannerRecommended by Élio Sucena based on reviews by Simon Fellous, alexandre leitão and 1 anonymous reviewerParasitoid wasps have developed different mechanisms to increase their parasitic success, usually at the expense of host survival (Fellowes and Godfray, 2000). Eggs of these insects are deposited inside the juvenile stages of their hosts, which in turn deploy several immune response strategies to eliminate or disable them (Yang et al., 2020). Drosophila melanogaster protects itself against parasitoid attacks through the production of specific elongated haemocytes called lamellocytes which form a capsule around the invading parasite (Lavine and Strand, 2002; Rizki and Rizki, 1992) and the subsequent activation of the phenol-oxidase cascade leading to the release of toxic radicals (Nappi et al., 1995). On the parasitoid side, robust responses have evolved to evade host immune defenses as for example the Drosophila-specific endoparasite Leptopilina boulardi, which releases venom during oviposition that modifies host behaviour (Varaldi et al., 2006) and inhibits encapsulation (Gueguen et al., 2011; Martinez et al., 2012).
References Cavigliasso, F., Mathé-Hubert, H., Gatti, J.-L., Colinet, D. and Poirié, M. (2021) Parasitic success and venom composition evolve upon specialization of parasitoid wasps to different host species. bioRxiv, 2020.10.24.353417, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.10.24.353417 Cavigliasso, F., Mathé-Hubert, H., Kremmer, L., Rebuf, C., Gatti, J.-L., Malausa, T., Colinet, D., Poiré, M. and Léne. (2019). Rapid and Differential Evolution of the Venom Composition of a Parasitoid Wasp Depending on the Host Strain. Toxins, 11(629). https://doi.org/10.3390/toxins11110629 Colinet, D., Deleury, E., Anselme, C., Cazes, D., Poulain, J., Azema-Dossat, C., Belghazi, M., Gatti, J. L. and Poirié, M. (2013). Extensive inter- and intraspecific venom variation in closely related parasites targeting the same host: The case of Leptopilina parasitoids of Drosophila. Insect Biochemistry and Molecular Biology, 43(7), 601–611. https://doi.org/10.1016/j.ibmb.2013.03.010 Colinet, D., Dubuffet, A., Cazes, D., Moreau, S., Drezen, J. M. and Poirié, M. (2009). A serpin from the parasitoid wasp Leptopilina boulardi targets the Drosophila phenoloxidase cascade. Developmental and Comparative Immunology, 33(5), 681–689. https://doi.org/10.1016/j.dci.2008.11.013 Fellowes, M. D. E. and Godfray, H. C. J. (2000). The evolutionary ecology of resistance to parasitoids by Drosophila. Heredity, 84(1), 1–8. https://doi.org/10.1046/j.1365-2540.2000.00685.x Gueguen, G., Rajwani, R., Paddibhatla, I., Morales, J. and Govind, S. (2011). VLPs of Leptopilina boulardi share biogenesis and overall stellate morphology with VLPs of the heterotoma clade. Virus Research, 160(1–2), 159–165. https://doi.org/10.1016/j.virusres.2011.06.005 Lavine, M. D. and Strand, M. R. (2002). Insect hemocytes and their role in immunity. Insect Biochemistry and Molecular Biology, 32(10), 1295–1309. https://doi.org/10.1016/S0965-1748(02)00092-9 Martinez, J., Duplouy, A., Woolfit, M., Vavre, F., O’Neill, S. L. and Varaldi, J. (2012). Influence of the virus LbFV and of Wolbachia in a host-parasitoid interaction. PloS One, 7(4), e35081. https://doi.org/10.1371/journal.pone.0035081 Nappi, A. J., Vass, E., Frey, F. and Carton, Y. (1995). Superoxide anion generation in Drosophila during melanotic encapsulation of parasites. European Journal of Cell Biology, 68(4), 450–456. Poirié, M., Colinet, D. and Gatti, J. L. (2014). Insights into function and evolution of parasitoid wasp venoms. Current Opinion in Insect Science, 6, 52–60. https://doi.org/10.1016/j.cois.2014.10.004 Rizki, T. M. and Rizki, R. M. (1992). Lamellocyte differentiation in Drosophila larvae parasitized by Leptopilina. Developmental and Comparative Immunology, 16(2–3), 103–110. https://doi.org/10.1016/0145-305X(92)90011-Z Schlenke, T. A., Morales, J., Govind, S. and Clark, A. G. (2007). Contrasting infection strategies in generalist and specialist wasp parasitoids of Drosophila melanogaster. PLoS Pathogens, 3(10), 1486–1501. https://doi.org/10.1371/journal.ppat.0030158 Varaldi, J., Petit, S., Boulétreau, M. and Fleury, F. (2006). The virus infecting the parasitoid Leptopilina boulardi exerts a specific action on superparasitism behaviour. Parasitology, 132(Pt 6), 747–756. https://doi.org/10.1017/S0031182006009930 Yang, L., Qiu, L., Fang, Q., Stanley, D. W. and Gong‐Yin, Y. (2020). Cellular and humoral immune interactions between Drosophila and its parasitoids. Insect Science. https://doi.org/10.1111/1744-7917.12863
| Parasitic success and venom composition evolve upon specialization of parasitoid wasps to different host species | Fanny Cavigliasso, Hugo Mathé-Hubert, Jean-Luc Gatti, Dominique Colinet, Marylène Poirié | <p>Female endoparasitoid wasps usually inject venom into hosts to suppress their immune response and ensure offspring development. However, the parasitoid’s ability to evolve towards increased success on a given host simultaneously with the evolut... | | Experimental Evolution, Species interactions | Élio Sucena | 2020-10-26 15:00:55 | ||
20 Nov 2023
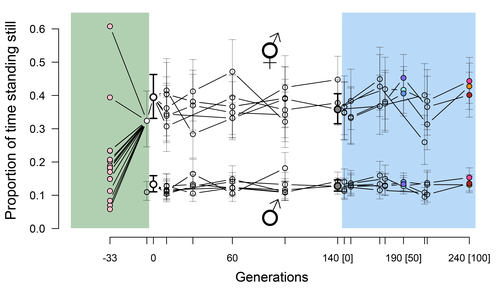
Phenotypic stasis with genetic divergencePhenotypic stasis despite genetic divergence and differentiation in Caenorhabditis elegans.Recommended by Frédéric Guillaume based on reviews by Benoit Pujol and Pedro Simões
Explaining long periods of evolutionary stasis, the absence of change in trait means over geological times, despite the existence of abundant genetic variation in most traits has challenged evolutionary theory since Darwin's theory of evolution by gradual modification (Estes & Arnold 2007). Stasis observed in contemporary populations is even more daunting since ample genetic variation is usually coupled with the detection of selection differentials (Kruuk et al. 2002, Morrissey et al. 2010). Moreover, rapid adaptation to environmental changes in contemporary populations, fuelled by standing genetic variation provides evidence that populations can quickly respond to an adaptive challenge. Explanations for evolutionary stasis usually invoke stabilizing selection as a main actor, whereby optimal trait values remain roughly constant over long periods of time despite small-scale environmental fluctuations. Genetic correlation among traits may also play a significant role in constraining evolutionary changes over long timescales (Schluter 1996). Yet, genetic constraints are rarely so strong as to completely annihilate genetic changes, and they may evolve. Patterns of genetic correlations among traits, as captured in estimates of the G-matrix of additive genetic co-variation, are subject to changes over generations under the action of drift, migration, or selection, among other causes (Arnold et al. 2008). Therefore, under the assumption of stabilizing selection on a set of traits, phenotypic stasis and genetic divergence in patterns of trait correlations may both be observed when selection on trait correlations is weak relative to its effect on trait means. Mallard et al. (2023) set out to test whether selection or drift may explain the divergence in genetic correlation among traits in experimental lines of the nematode Caenorhabditis elegans and whether stabilizing selection may be a driver of phenotypic stasis. To do so, they analyzed the evolution of locomotion behavior traits over 100 generations of lab evolution in a constant and homogeneous environment after 140 generations of domestication from a largely differentiated set of founder populations. The locomotion traits were transition rates between movement states and direction (still, forward or backward movement). They could estimate the traits' broad-sense G-matrix in three populations at two generations (50 and 100), and in the ancestral mixed population. Similarly, they estimated the shape of the selection surface by regressing locomotion behavior on fertility. Armed with both G-matrix and surface estimates, they could test whether the G's orientation matched selection's orientation and whether changes in G were constrained by selection. They found stasis in trait mean over 100 generations but divergence in the amount and orientation of the genetic variation of the traits relative to the ancestral population. The selected populations changed orientation of their G-matrices and lost genetic variation during the experiment in agreement with a model of genetic drift on quantitative traits. Their estimates of selection also point to mostly stabilizing selection on trait combinations with weak evidence of disruptive selection, suggesting a saddle-shaped selection surface. The evolutionary responses of the experimental populations were mostly consistent with small differentiation in the shape of G-matrices during the 100 generations of stabilizing selection. Mallard et al. (2023) conclude that phenotypic stasis was maintained by stabilizing selection and drift in their experiment. They argue that their findings are consistent with a "table-top mountain" model of stabilizing selection, whereby the population is allowed some wiggle room around the trait optimum, leaving space for random fluctuations of trait variation, and especially trait co-variation. The model is an interesting solution that might explain how stasis can be maintained over contemporary times while allowing for random differentiation of trait genetic co-variation. Whether such differentiation can then lead to future evolutionary divergence once replicated populations adapt to a new environment is an interesting idea to follow. References Arnold, S. J., Bürger, R., Hohenlohe, P. A., Ajie, B. C. and Jones, A. G. 2008. Understanding the evolution and stability of the G-matrix. Evolution 62(10): 2451-2461. | Phenotypic stasis with genetic divergence | François Mallard, Luke Noble, Thiago Guzella, Bruno Afonso, Charles F. Baer, Henrique Teotónio | <p style="text-align: justify;">Whether or not genetic divergence in the short-term of tens to hundreds of generations is compatible with phenotypic stasis remains a relatively unexplored problem. We evolved predominantly outcrossing, genetically ... | | Adaptation, Behavior & Social Evolution, Experimental Evolution, Quantitative Genetics | Frédéric Guillaume | 2022-09-01 14:32:53 | ||
09 Feb 2018
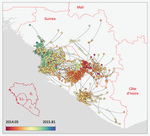
Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreakSimulating the effect of public health interventions using dated virus sequences and geographical dataRecommended by Samuel Alizon based on reviews by Christian Althaus, Chris Wymant and 1 anonymous reviewer
Perhaps because of its deadliness, the 2013-2016 Ebola Virus (EBOV) epidemics in West-Africa has led to unprecedented publication and sharing of full virus genome sequences. This was both rapid (90 full genomes were shared within weeks [1]) and important (more than 1500 full genomes have been released overall [2]). Furthermore, the availability of the metadata (especially GPS location) has led to depth analyses of the geographical spread of the epidemics [3]. References [1] Gire et al. 2014. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345: 1369–1372. doi: 10.1126/science.1259657. | Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreak | Simon Dellicour, Guy Baele, Gytis Dudas, Nuno R. Faria, Oliver G. Pybus, Marc A. Suchard, Andrew Rambaut, Philippe Lemey | <p>This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (https://doi.org/10.24072/pci.evolbiol.100046). The recent Ebola virus (EBOV) outbreak in West Africa witnessed considerable efforts to obtain viral genom... | | Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Samuel Alizon | 2017-09-30 13:49:57 | ||
11 Mar 2020
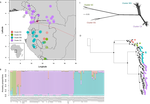
Phylogenomic approaches reveal how a climatic inversion and glacial refugia shape patterns of diversity in an African rain forest tree speciesRemarkable insights into processes shaping African tropical tree diversityRecommended by Michael Pirie based on reviews by Miguel de Navascués, Lars Chatrou and Oscar Vargas
Tropical biodiversity is immense, under enormous threat, and yet still poorly understood. Global climatic breakdown and habitat destruction are impacting on and removing this diversity before we can understand how the biota responds to such changes, or even fully appreciate what we are losing [1]. This is particularly the case for woody shrubs and trees [2] and for the flora of tropical Africa [3]. Helmstetter et al. [4] have taken a significant step to improve our understanding of African tropical tree diversity in the context of past climatic change. They have done so by means of a remarkably in-depth analysis of one species of the tropical plant family Annonaceae: Annickia affinis [5]. A. affinis shows a distribution pattern in Africa found in various plant (but interestingly not animal) groups: a discontinuity between north and south of the equator [6]. There is no obvious physical barrier to cause this discontinuity, but it does correspond with present day distinct northern and southern rainy seasons. Various explanations have been proposed for this discontinuity, set out as hypotheses to be tested in this paper: climatic fluctuations resulting in changes in plant distributions in the Pleistocene, or differences in flowering times or in ecological niche between northerly and southerly populations. These explanations are not mutually exclusive, but they can be tested using phylogenetic inference – if you can sample variable enough sequence data from enough individuals – complemented with analysis of ecological niches and traits. Using targeted sequence capture, the authors amassed a dataset representing 351 nuclear markers for 112 individuals of A. affinis. This dataset is impressive for a number of reasons: First, sampling such a species across such a wide range in tropical Africa presents numerous challenges of itself. Second, the technical achievement of using this still relatively new sequencing technique with a custom set of baits designed specifically for this plant family [7] is also considerable. The result is a volume of data that just a few years ago would not have been feasible to collect, and which now offers the possibility to meaningfully analyse DNA sequence variation within a species across numerous independent loci of the nuclear genome. This is the future of our research field, and the authors have ably demonstrated some of its possibilities. Using this data, they performed on the one hand different population genetic clustering approaches, and on the other, different phylogenetic inference methods. I would draw attention to their use and comparison of coalescence and network-based approaches, which can account for the differences between gene trees that might be expected between populations of a single species. The results revealed four clades and a consistent sequence of divergences between them. The authors inferred past shifts in geographic range (using a continuous state phylogeographic model), depicting a biogeographic scenario involving a dispersal north over the north/south discontinuity; and demographic history, inferring in some (but not all) lineages increases in effective population size around the time of the last glacial maximum, suggestive of expansion from refugia. Using georeferenced specimen data, they compared ecological niches between populations, discovering that overlap was indeed smallest comparing north to south. Just the phenology results were effectively inconclusive: far better data on flowering times is needed than can currently be harvested from digitised herbarium specimens. Overall, the results add to the body of evidence for the impact of Pleistocene climatic changes on population structure, and for niche differences contributing to the present day north/south discontinuity. However, they also paint a complex picture of idiosyncratic lineage-specific responses, even within a single species. With the increasing accessibility of the techniques used here we can look forward to more such detailed analyses of independent clades necessary to test and to expand on these conclusions, better to understand the nature of our tropical plant diversity while there is still opportunity to preserve it for future generations. References [1] Mace, G. M., Gittleman, J. L., and Purvis, A. (2003). Preserving the Tree of Life. Science, 300(5626), 1707–1709. doi: 10.1126/science.1085510 | Phylogenomic approaches reveal how a climatic inversion and glacial refugia shape patterns of diversity in an African rain forest tree species | Andrew J. Helmstetter, Biowa E. N. Amoussou, Kevin Bethune, Narcisse G. Kandem, Romain Glèlè Kakaï, Bonaventure Sonké, Thomas L. P. Couvreur | <p>The world’s second largest expanse of tropical rain forest is in Central Africa and it harbours enormous species diversity. Population genetic studies have consistently revealed significant structure across central African rain forest plants, i... | | Evolutionary Dynamics, Phylogeography & Biogeography | Michael Pirie | 2019-10-29 15:19:36 |




