
Latest recommendations

| Id | Title▲ | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
13 Dec 2016

POSTPRINT
Repeated replacements of an intrabacterial symbiont in the tripartite nested mealybug symbiosisObligate dependence does not preclude changing partners in a Russian dolls symbiotic systemRecommended by Emmanuelle Jousselin and Fabrice VavreSymbiotic associations with bacterial partners have facilitated important evolutionary transitions in the life histories of eukaryotes. For instance, many insects have established long-term interactions with intracellular bacteria that provide them with essential nutrients lacking in their diet. However, despite the high level of interdependency among organisms involved in endosymbiotic systems, examples of symbiont replacements along the evolutionary history of insect hosts are numerous.
In their paper, Husnik and McCutcheon [1] test the stability of symbiotic systems in a particularly imbricated Russian-doll type interaction, where one bacterium lives insides another bacterium, which itself lives inside insect cells. For their study, they chose representative species of mealybugs (Pseudococcidae), a species rich group of sap-feeding insects that hosts diverse and complex symbiotic systems. In species of the subfamily Pseudococcinae, data published so far suggest that the primary symbiont, a ß-proteobacterium named Tremblaya princeps, is supplemented by a second bacterial symbiont (a ϒ-proteobaterium) that lives within its cytoplasm; both participate to the metabolic pathways that provide essential amino acids and vitamins to their hosts. Here, Husnik and McCutcheon generate host and endosymbiont genome data for five phylogenetically divergent species of Pseudococcinae in order to better understand: 1) the evolutionary history of the symbiotic associations; 2) the metabolic roles of each partner, 3) the timing and origin of Horizontal Gene Transfers (HGT) between the hosts and their symbionts. Reference [1] Husnik F., McCutcheon JP. 2016. Repeated replacements of an intrabacterial symbiont in the tripartite nested mealybug symbiosis. PNAS 113: E5416-E5424. doi: 10.1073/pnas.1603910113 | Repeated replacements of an intrabacterial symbiont in the tripartite nested mealybug symbiosis | Husnik F, McCutcheon JP | Stable endosymbiosis of a bacterium into a host cell promotes cellular and genomic complexity. The mealybug *Planococcus citri* has two bacterial endosymbionts with an unusual nested arrangement: the γ-proteobacterium *Moranella endobia* lives in ... | | Phylogenetics / Phylogenomics, Species interactions | Emmanuelle Jousselin | 2016-12-13 14:27:09 | ||
31 Jul 2017

Selection on morphological traits and fluctuating asymmetry by a fungal parasite in the yellow dung flyParasite-mediated selection promotes small body size in yellow dung fliesRecommended by Rodrigo Medel based on reviews by Rodrigo Medel and 1 anonymous reviewerBody size has long been considered as one of the most important organismic traits influencing demographical processes, population size, and evolution of life history strategies [1, 2]. While many studies have reported a selective advantage of large body size, the forces that determine small-sized organisms are less known, and reports of negative selection coefficients on body size are almost absent at present. This lack of knowledge is unfortunate as climate change and energy demands in stressful environments, among other factors, may produce new selection scenarios and unexpected selection surfaces [3]. In this manuscript, Blanckenhorn [4] reports on a potential explanation for the surprising 10% body size decrease observed in a Swiss population of yellow dung flies during 1993 - 2009. The author took advantage of a fungus outbreak in 2002 to assess the putative role of the fungus Entomopthora scatophagae, a specific parasite of adult yellow dung flies, as selective force acting upon host body size. His findings indicate that, as expected by sexual selection theory, large males experience a mating advantage. However, this positive sexual selection is opposed by a strong negative selection on male and female body size through the viability fitness component. This study provides the first evidence of parasite-mediated disadvantage of large adult body size in the field. While further experimental work is needed to elucidate the exact causes of body size reduction in the population, the author proposes a variation of the trade-off hypothesis raised by Rantala & Roff [5] that large-sized individuals face an immunity cost due to their high absolute energy demands in stressful environments. References [1] Peters RH. 1983. The ecological implications of body size. Cambridge University Press, Cambridge. [2] Schmidt-Nielsen K. 1984. Scaling: why is animal size so important? Cambridge University Press, Cambridge. [3] Ohlberger J. 2013. Climate warming and ectotherm body size: from individual physiology to community ecology. Functional Ecology 27: 991-1001. doi: 10.1111/1365-2435.12098 [4] Blanckenhorn WU. 2017. Selection on morphological traits and fluctuating asymmetry by a fungal parasite in the yellow dung fly. bioRxiv 136325, ver. 2 of 29th June 2017. doi: 10.1101/136325 [5] Rantala MJ & Roff DA. 2005. An analysis of trade-offs in immune function, body size and development time in the Mediterranean field cricket, Gryllus bimaculatus. Functional Ecology 19: 323-330. doi: 10.1111/j.1365-2435.2005.00979.x | Selection on morphological traits and fluctuating asymmetry by a fungal parasite in the yellow dung fly | Wolf U. Blanckenhorn | Evidence for selective disadvantages of large body size remains scarce in general. Previous phenomenological studies of the yellow dung fly *Scathophaga stercoraria* have demonstrated strong positive sexual and fecundity selection on male and fema... | | Behavior & Social Evolution, Evolutionary Ecology, Life History, Sexual Selection | Rodrigo Medel | Rodrigo Medel | 2017-05-10 11:16:26 | |
04 Aug 2023

Sensitive windows for within- and trans-generational plasticity of anti-predator defencesSensitive windows for phenotypic plasticity within and across generations; where empirical results do not meet the theory but open a world of possibilitiesRecommended by Benoit Pujol based on reviews by David Murray-Stoker, Timothée Bonnet and Willem FrankenhuisIt is easy to define phenotypic plasticity as a mechanism by which traits change in response to a modification of the environment. Many complex mechanisms are nevertheless involved with plastic responses, their strength, and stability (e.g., reliability of cues, type of exposure, genetic expression, epigenetics). It is rather intuitive to think that environmental cues perceived at different stages of development will logically drive different phenotypic responses (Fawcett and Frankenhuis 2015). However, it has proven challenging to try and explain, or model how and why different effects are caused by similar cues experienced at different developmental or life stages (Walasek et al. 2022). The impact of these ‘sensitive windows’ on the stability of plastic responses within or across generations remains unclear. In their paper entitled “Sensitive windows for within- and trans-generational plasticity of anti-predator defences”, Tariel-Adam (2023) address this question. In this paper, Tariel et al. acknowledge the current state of the art, i.e., that some traits influenced by the environment at early life stages become fixed later in life (Snell-Rood et al. 2015) and that sensitive windows are therefore more likely to be observed during early stages of development. Constructive exchanges with the reviewers illustrated that Tariel et al. presented a clear picture of the knowledge on sensitive windows from a conceptual and a mechanistic perspective, thereby providing their study with a strong and elegant rationale. Tariel et al. outlined that little is known about the significance of this scenario when it comes to transgenerational plasticity. Theory predicts that exposure late in the life of parents should be more likely to drive transgenerational plasticity because the cue perceived by parents is more likely to be reliable if time between parental exposure and offspring expression is short (McNamara et al. 2016). I would argue that although sensible, this scenario is likely oversimplifying the complexity of evolutionary, ecological, and inheritance mechanisms at play (Danchin et al. 2018). Tariel-Adam et al. (2023) point out in their paper how the absence of experimental results limits our understanding of the evolutionary and adaptive significance of transgenerational plasticity and decided to address this broad question. Tariel-Adam et al. (2023) used the context of predator-prey interactions, which is a powerful framework to evaluate the temporality of predator cues and prey responses within and across generations (Sentis et al. 2018). They conducted a very elegant experiment whereby two generations of freshwater snails Physa acuta were exposed to crayfish predator cues at different developmental windows. They triggered the within-generation phenotypic plastic response of inducible defences (e.g., shell thickness) and identified sensitive windows as to evaluate their role in within-generation phenotypic plasticity versus transgenerational plasticity. They used different linear models, which lead to constructive exchanges with reviewers, and between reviewers, well trained on these approaches, in particular on effect sizes, that improved the paper by pushing the discussion all the way towards a consensus. Tariel-Adam et al. (2023) results showed that the phenotypic plastic response of different traits was associated with different sensitive windows. Although early-life development was confirmed to be a sensitive window, it was far from being the only developmental stage driving within-generation plastic responses of defence traits. This finding contributes to change our views on plasticity because where theoretical models predict early- and late-life sensitive windows, empirical results gathered here present a more continuous opportunity for sensitive windows over the lifetime of freshwater snails. This is likely because multifactorial mechanisms drive the reliability and adaptive significance of predator cues. To me, this paper most original contribution lies probably in the empirical investigation of sensitive windows underlying transgenerational plasticity. Their finding implies mechanistic ties between sensitive windows driving within-generation and transgenerational plasticity for some traits, but they also shed light on the possible independence of these processes. Although one may be disheartened by these findings illustrating the ability of nature to combine complex mechanisms in order to produce somewhat unpredictable scenarios, one can only find that this unlimited range of phenotypic plasticity scenarios is a wonder to investigate because much remains to be understood. As mentioned in the conclusion of the paper, the opportunity for sensitive windows to drive such a range of plastic responses may also be an opportunity for organisms to adapt to a wide range of environmental demands. References Danchin E, A Pocheville, O Rey, B Pujol, and S Blanchet (2019). Epigenetically facilitated mutational assimilation: epigenetics as a hub within the inclusive evolutionary synthesis. Biological Reviews, 94: 259-282. https://doi.org/10.1111/brv.12453 Fawcett TW, and WE Frankenhuis (2015). Adaptive Explanations for Sensitive Windows in Development. Frontiers in Zoology 12, S3. https://doi.org/10.1186/1742-9994-12-S1-S3 McNamara JM, SRX Dall, P Hammerstein, and O Leimar (2016). Detection vs. Selection: Integration of Genetic, Epigenetic and Environmental Cues in Fluctuating Environments. Ecology Letters 19, 1267–1276. https://doi.org/10.1111/ele.12663 Sentis A, R Bertram, N Dardenne, et al. (2018). Evolution without standing genetic variation: change in transgenerational plastic response under persistent predation pressure. Heredity 121, 266–281. https://doi.org/10.1038/s41437-018-0108-8 Snell-Rood EC, EM Swanson, and RL Young (2015). Life History as a Constraint on Plasticity: Developmental Timing Is Correlated with Phenotypic Variation in Birds. Heredity 115, 379–388. https://doi.org/10.1038/hdy.2015.47 Tariel-Adam J, E Luquet, and S Plénet (2023). Sensitive windows for within- and trans-generational plasticity of anti-predator defences. OSF preprints, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.31219/osf.io/mr8hu Walasek N, WE Frankenhuis, and K Panchanathan (2022). An Evolutionary Model of Sensitive Periods When the Reliability of Cues Varies across Ontogeny. Behavioral Ecology 33, 101–114. https://doi.org/10.1093/beheco/arab113 | Sensitive windows for within- and trans-generational plasticity of anti-predator defences | Juliette Tariel-Adam; Émilien Luquet; Sandrine Plénet | <p>Transgenerational plasticity could be an important mechanism for adaptation to variable environments in addition to within-generational plasticity. But its potential for adaptation may be restricted to specific developmental windows that are hi... | | Adaptation, Evolutionary Ecology, Phenotypic Plasticity | Benoit Pujol | 2022-11-14 08:08:27 | ||
16 Jun 2022

Sensory plasticity in a socially plastic beeTaking advantage of facultative sociality in sweat bees to study the developmental plasticity of antennal sense organs and its association with social phenotypeRecommended by Nadia Aubin-Horth based on reviews by Michael D Greenfield, Sylvia Anton and Lluís Socias-MartínezThe study of the evolution of sociality is closely associated with the study of the evolution of sensory systems. Indeed, group life and sociality necessitate that individuals recognize each other and detect outsiders, as seen in eusocial insects such as Hymenoptera. While we know that antennal sense organs that are involved in olfactory perception are found in greater densities in social species of that group compared to solitary hymenopterans, whether this among-species correlation represents the consequence of social evolution leading to sensory evolution, or the opposite, is still questioned. Knowing more about how sociality and sensory abilities covary within a species would help us understand the evolutionary sequence. Studying a species that shows social plasticity, that is facultatively social, would further allow disentangling the cause and consequence of social evolution and sensory systems and the implication of plasticity in the process. Boulton and Field (2022) studied a species of sweat bee that shows social plasticity, Halictus rubicundus. They studied populations at different latitudes in Great Britain: populations in the North are solitary, while populations in the south often show sociality, as they face a longer and warmer growing season, leading to the opportunity for two generations in a single year, a pre-condition for the presence of workers provisioning for the (second) brood. Using scanning electron microscope imaging, the authors compared the density of antennal sensilla types in these different populations (north, mid-latitude, south) to test for an association between sociality and olfactory perception capacities. They counted three distinct types of antennal sensilla: olfactory plates, olfactory hairs, and thermos/hygro-receptive pores, used to detect humidity, temperature and CO2. In addition, they took advantage of facultative sociality in this species by transplanting individuals from a northern population (solitary) to a southern location (where conditions favour sociality), to study how social plasticity is reflected (or not) in the density of antennal sensilla types. They tested the prediction that olfactory sensilla density is also developmentally plastic in this species. Their results show that antennal sensilla counts differ between the 3 studied regions (north, mid-latitude, south), but not as predicted. Individuals in the southern population were not significantly different from the mid-latitude and northern ones in their count of olfactory plates and they had less, not more, thermos/hygro receptors than mid-latitude and northern individuals. Furthermore, mid-latitude individuals had more olfactory hairs than the ones from the northern population and did not differ from southern ones. The prediction was that the individuals expressing sociality would have the highest count of these olfactory hairs. This unpredicted pattern based on the latitude of sampling sites may be due to the effect of temperature during development, which was higher in the mid-latitude site than in the southern one. It could also be the result of a genotype-by-environment interaction, where the mid-latitude population has a different developmental response to temperature compared to the other populations, a difference that is genetically determined (a different “reaction norm”). Reciprocal transplant experiments coupled with temperature measurements directly on site would provide interesting information to help further dissect this intriguing pattern. Interestingly, where a sweat bee developed had a significant effect on their antennal sensilla counts: individuals originating from the North that developed in the south after transplantation had significantly more olfactory hairs on their antenna than individuals from the same Northern population that developed in the North. This is in accordance with the prediction that the characteristics of sensory organs can also be plastic. However, there was no difference in antennal characteristics depending on whether these transplanted bees became solitary or expressed the social phenotype (foundress or worker). This result further supports the hypothesis that temperature affects development in this species and that these sensory characteristics are also plastic, although independently of sociality. Overall, the work of Boulton and Field underscores the importance of including phenotypic plasticity in the study of the evolution of social behaviour and provides a robust and fruitful model system to explore this further. References Boulton RA, Field J (2022) Sensory plasticity in a socially plastic bee. bioRxiv, 2022.01.29.478030, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.01.29.478030 | Sensory plasticity in a socially plastic bee | Rebecca A Boulton, Jeremy Field | <p style="text-align: justify;">The social Hymenoptera have contributed much to our understanding of the evolution of sensory systems. Attention has focussed chiefly on how sociality and sensory systems have evolved together. In the Hymenoptera, t... | | Behavior & Social Evolution, Evolutionary Ecology, Phenotypic Plasticity | Nadia Aubin-Horth | 2022-02-02 11:34:49 | ||
13 Dec 2018
Separate the wheat from the chaff: genomic analysis of local adaptation in the red coral Corallium rubrumPros and Cons of local adaptation scansRecommended by Guillaume Achaz based on reviews by Lucas Gonçalves da Silva and 1 anonymous reviewerThe preprint by Pratlong et al. [1] is a well thought quest for genomic regions involved in local adaptation to depth in a species a red coral living the Mediterranean Sea. It first describes a pattern of structuration and then attempts to find candidate genes involved in local adaptation by contrasting deep with shallow populations. Although the pattern of structuration is clear and meaningful, the candidate genomic regions involved in local adaptation remain to be confirmed. Two external reviewers and myself found this preprint particularly interesting regarding the right-mindedness of the authors in front of the difficulties they encounter during their experiments. The discussions on the pros and cons of the approach are very sound and can be easily exported to a large number of studies that hunt for local adaptation. In this sense, the lessons one can learn by reading this well documented manuscript are certainly valuable for a wide range of evolutionary biologists. References [1] Pratlong, M., Haguenauer, A., Brener, K., Mitta, G., Toulza, E., Garrabou, J., Bensoussan, N., Pontarotti P., & Aurelle, D. (2018). Separate the wheat from the chaff: genomic scan for local adaptation in the red coral Corallium rubrum. bioRxiv, 306456, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/306456 | Separate the wheat from the chaff: genomic analysis of local adaptation in the red coral Corallium rubrum | Pratlong M, Haguenauer A, Brener K, Mitta G, Toulza E, Garrabou J, Bensoussan N, Pontarotti P, Aurelle D | <p>Genomic data allow an in-depth and renewed study of local adaptation. The red coral (Corallium rubrum, Cnidaria) is a highly genetically structured species and a promising model for the study of adaptive processes along an environmental gradien... | | Adaptation, Population Genetics / Genomics | Guillaume Achaz | 2018-04-24 11:27:40 | ||
08 Aug 2018

Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutationsInbreeding compensates for reduced sexual selection in purging deleterious mutationsRecommended by Charles Baer based on reviews by 2 anonymous reviewersTwo evolutionary processes have been shown in theory to enhance the effects of natural selection in purging deleterious mutations from a population (here ""natural"" selection is defined as ""selection other than sexual selection""). First, inbreeding, especially self-fertilization, facilitates the removal of deleterious recessive alleles, the effects of which are largely hidden from selection in heterozygotes when mating is random. Second, sexual selection can facilitate the removal of deleterious alleles of arbitrary dominance, with little or no demographic cost, provided that deleterious effects are greater in males than in females (""genic capture""). Inbreeding (especially selfing) and sexual selection are often negatively correlated in nature. Empirical tests of the role of sexual selection in purging deleterious mutations have been inconsistent, potentially due to the positive relationship between sexual selection and intersexual genetic conflict. References [1] Noël, E., Fruitet, E., Lelaurin, D., Bonel, N., Segard, A., Sarda, V., Jarne, P., & David P. (2018). Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutations. bioRxiv, 273367, ver. 3 recommended and peer-reviewed by PCI Evol Biol. doi: 10.1101/273367 | Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutations | E. Noël, E. Fruitet, D. Lelaurin, N. Bonel, A. Ségard, V. Sarda, P. Jarne and P. David | <p>This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (https://dx.doi.org/10.24072/pci.evolbiol.100055). Theory and empirical data showed that two processes can boost selection against deleterious mutations, ... | | Adaptation, Experimental Evolution, Reproduction and Sex, Sexual Selection | Charles Baer | Anonymous | 2018-03-01 08:12:37 | |
16 Dec 2020

Shifts from pulled to pushed range expansions caused by reduction of landscape connectivityThe push and pull between theory and data in understanding the dynamics of invasionRecommended by Ben Phillips based on reviews by Laura Naslund and 2 anonymous reviewersExciting times are afoot for those of us interested in the ecology and evolution of invasive populations. Recent years have seen evolutionary process woven firmly into our understanding of invasions (Miller et al. 2020). This integration has inspired a welter of empirical and theoretical work. We have moved from field observations and verbal models to replicate experiments and sophisticated mathematical models. Progress has been rapid, and we have seen science at its best; an intimate discussion between theory and data. References Bîrzu, G., Matin, S., Hallatschek, O., and Korolev, K. S. (2019). Genetic drift in range expansions is very sensitive to density dependence in dispersal and growth. Ecology Letters, 22(11), 1817-1827. doi: https://doi.org/10.1111/ele.13364 | Shifts from pulled to pushed range expansions caused by reduction of landscape connectivity | Maxime Dahirel, Aline Bertin, Marjorie Haond, Aurélie Blin, Eric Lombaert, Vincent Calcagno, Simon Fellous, Ludovic Mailleret, Thibaut Malausa, Elodie Vercken | <p>Range expansions are key processes shaping the distribution of species; their ecological and evolutionary dynamics have become especially relevant today, as human influence reshapes ecosystems worldwide. Many attempts to explain and predict ran... | | Evolutionary Applications, Evolutionary Dynamics, Evolutionary Ecology, Experimental Evolution, Phylogeography & Biogeography | Ben Phillips | 2020-08-04 12:51:56 | ||
04 Mar 2021
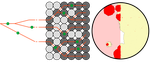
Simulation of bacterial populations with SLiMSimulating bacterial evolution forward-in-timeRecommended by Frederic Bertels based on reviews by 3 anonymous reviewersJean Cury and colleagues (2021) have developed a protocol to simulate bacterial evolution in SLiM. In contrast to existing methods that depend on the coalescent, SLiM simulates evolution forward in time. SLiM has, up to now, mostly been used to simulate the evolution of eukaryotes (Haller and Messer 2019), but has been adapted here to simulate evolution in bacteria. Forward-in-time simulations are usually computationally very costly. To circumvent this issue, bacterial population sizes are scaled down. One would now expect results to become inaccurate, however, Cury et al. show that scaled-down forwards simulations provide very accurate results (similar to those provided by coalescent simulators) that are consistent with theoretical expectations. Simulations were analyzed and compared to existing methods in simple and slightly more complex scenarios where recombination affects evolution. In all scenarios, simulation results from coalescent methods (fastSimBac (De Maio and Wilson 2017), ms (Hudson 2002)) and scaled-down forwards simulations were very similar, which is very good news indeed. A biologist not aware of the complexities of forwards, backwards simulations and the coalescent, might now naïvely ask why another simulation method is needed if existing methods perform just as well. To address this question the manuscript closes with a very neat example of what exactly is possible with forwards simulations that cannot be achieved using existing methods. The situation modeled is the growth and evolution of a set of 50 bacteria that are randomly distributed on a petri dish. One side of the petri dish is covered in an antibiotic the other is antibiotic-free. Over time, the bacteria grow and acquire antibiotic resistance mutations until the entire artificial petri dish is covered with a bacterial lawn. This simulation demonstrates that it is possible to simulate extremely complex (e.g. real world) scenarios to, for example, assess whether certain phenomena are expected with our current understanding of bacterial evolution, or whether there are additional forces that need to be taken into account. Hence, forwards simulators could significantly help us to understand what current models can and cannot explain in evolutionary biology.
References Cury J, Haller BC, Achaz G, Jay F (2021) Simulation of bacterial populations with SLiM. bioRxiv, 2020.09.28.316869, version 5 peer-reviewed and recommended by Peer community in Evolutionary Biology. https://doi.org/10.1101/2020.09.28.316869 De Maio N, Wilson DJ (2017) The Bacterial Sequential Markov Coalescent. Genetics, 206, 333–343. https://doi.org/10.1534/genetics.116.198796 Haller BC, Messer PW (2019) SLiM 3: Forward Genetic Simulations Beyond the Wright–Fisher Model. Molecular Biology and Evolution, 36, 632–637. https://doi.org/10.1093/molbev/msy228 Hudson RR (2002) Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics, 18, 337–338. https://doi.org/10.1093/bioinformatics/18.2.337 | Simulation of bacterial populations with SLiM | Jean Cury, Benjamin C. Haller, Guillaume Achaz, and Flora Jay | <p>Simulation of genomic data is a key tool in population genetics, yet, to date, there is no forward-in-time simulator of bacterial populations that is both computationally efficient and adaptable to a wide range of scenarios. Here we demonstrate... | | Bioinformatics & Computational Biology, Population Genetics / Genomics | Frederic Bertels | 2020-10-02 19:03:42 | ||
04 Mar 2024
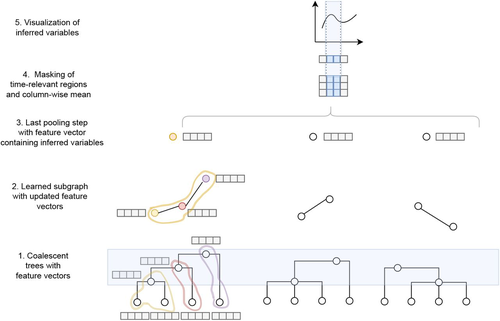
Simultaneous Inference of Past Demography and Selection from the Ancestral Recombination Graph under the Beta CoalescentBeyond the standard coalescent: demographic inference with complete genomes and graph neural networks under the beta coalescentRecommended by Julien Yann Dutheil based on reviews by 2 anonymous reviewers based on reviews by 2 anonymous reviewers
Modelling the evolution of complete genome sequences in populations requires accounting for the recombination process, as a single tree can no longer describe the underlying genealogy. The sequentially Markov coalescent (SMC, McVean and Cardin 2005; Marjoram and Wall 2006) approximates the standard coalescent with recombination process and permits estimating population genetic parameters (e.g., population sizes, recombination rates) using population genomic datasets. As such datasets become available for an increasing number of species, more fine-tuned models are needed to encompass the diversity of life cycles of organisms beyond the model species on which most methods have been benchmarked. The work by Korfmann et al. (Korfmann et al. 2024) represents a significant step forward as it accounts for multiple mergers in SMC models. Multiple merger models account for simultaneous coalescence events so that more than two lineages find a common ancestor in a given generation. This feature is not allowed in standard coalescent models and may result from selection or skewed offspring distributions, conditions likely met by a broad range of species, particularly microbial. Yet, this work goes beyond extending the SMC, as it introduces several methodological innovations. The "classical" SMC-based inference approaches rely on hidden Markov models to compute the likelihood of the data while efficiently integrating over the possible ancestral recombination graphs (ARG). Following other recent works (e.g. Gattepaille et al. 2016), Korfmann et al. propose to separate the ARG inference from model parameter estimation under maximum likelihood (ML). They introduce a procedure where the ARG is first reconstructed from the data and then taken as input in the model fitting step. While this approach does not permit accounting for the uncertainty in the ARG reconstruction (which is typically large), it potentially allows for the extraction of more information from the ARG, such as the occurrence of multiple merging events. Going away from maximum likelihood inference, the authors trained a graph neural network (GNN) on simulated ARGs, introducing a new, flexible way to estimate population genomic parameters. The authors used simulations under a beta-coalescent model with diverse demographic scenarios and showed that the ML and GNN approaches introduced can reliably recover the simulated parameter values. They further show that when the true ARG is given as input, the GNN outperforms the ML approach, demonstrating its promising power as ARG reconstruction methods improve. In particular, they showed that trained GNNs can disentangle the effects of selective sweeps and skewed offspring distributions while inferring past population size changes. This work paves the way for new, exciting applications, though many questions must be answered. How frequent are multiple mergers? As the authors showed that these events "erase" the record of past demographic events, how many genomes are needed to conduct reliable inference, and can the methods computationally cope with the resulting (potentially large) amounts of required data? This is particularly intriguing as micro-organisms, prone to strong selection and skewed offspring distributions, also tend to carry smaller genomes. References Gattepaille L, Günther T, Jakobsson M. 2016. Inferring Past Effective Population Size from Distributions of Coalescent Times. Genetics 204:1191-1206. | Simultaneous Inference of Past Demography and Selection from the Ancestral Recombination Graph under the Beta Coalescent | Kevin Korfmann, Thibaut Sellinger, Fabian Freund, Matteo Fumagalli, Aurélien Tellier | <p style="text-align: justify;">The reproductive mechanism of a species is a key driver of genome evolution. The standard Wright-Fisher model for the reproduction of individuals in a population assumes that each individual produces a number of off... | | Adaptation, Bioinformatics & Computational Biology, Evolutionary Applications, Evolutionary Theory, Life History, Population Genetics / Genomics | Julien Yann Dutheil | 2023-07-31 13:11:22 | ||
30 May 2023
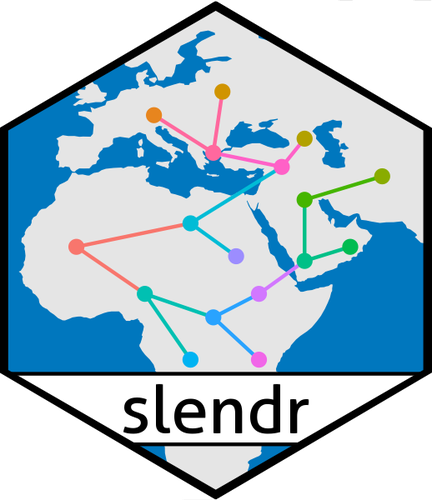
slendr: a framework for spatio-temporal population genomic simulations on geographic landscapesA new powerful tool to easily encode the geo-spatial dimension in population genetics simulationsRecommended by Emiliano Trucchi based on reviews by Liisa Loog and 2 anonymous reviewers
Models explaining the evolutionary processes operating in living beings are often impossible to test in the real world. This is mainly because of the long time (i.e., the number of generations) which is necessary for evolution to unfold. In addition, any such experiment would require a large number of individuals and, more importantly, many replicates to account for the inherent variance of the evolutionary processes under investigation. Only organisms with fast generation times and favourable rearing conditions can be used to explicitly test for specific evolutionary hypotheses. Computer simulations have filled this gap, revolutionising experimental testing in evolutionary biology by integrating genetic models into complex population dynamics, which can be run for (potentially) any length of time. Without going into an extensive description of the many available approaches for population genetics simulations (an exhaustive review can be found in Hoban et al 2012), three main aspects are, in my opinion, important for categorising and choosing one simulation approach over another. The first concerns the basic distinction between coalescent-based and individual-based simulators: the former being an efficient approach, which simulates back in time the coalescence events of a sample of homologous DNA fragments, while the latter is a more computationally intensive approach where all of the individuals (and their underlying genetic/genomic features) in the population are simulated forward-in-time, generation after generation. The second aspect concerns the simulation of natural selection. Although natural selection can be integrated into backward-in-time simulations, it is more realistically implemented as individual-based fitness in forward-in-time simulators. The third point, which has been often overlooked in evolutionary simulations, is about the possibility to design a simulation scenario where individuals and populations can exploit a physical (geographical) space. Amongst the coalescent-based simulators, SPLATCHE (Currat et al 2004), and its derivatives, is one of the few simulation tools deploying the coalescence process in sub-demes which are all connected by migration, thus getting as close as possible to a spatially-explicit population. On the other hand, individual-based simulators, whose development followed the increasing power of computational machines, offer a great opportunity to include spatio-temporal dynamics within a genomic simulation model. One of the most realistic and efficient individual-based forward-in-time simulators available is SLiM (Haller and Messer 2017), which allows users to implement simulations in arbitrarily complex spaces. Here, the more challenging part is encoding the spatially-explicit scenarios using the SLiM-specific EIDOS language. The new R package slendr (Petr et al 2022) offers a practical solution to this issue. By wrapping different tools into a well-known scripting language, slendr allows the design of spatiotemporal simulation scenarios which can be directly executed in the individual-based SLiM simulator, and the output stored with modern tree-sequence analysis tools (tskit; Kellerer et al 2018). Alternatively, simulations of non-spatial models can be run using a coalescent-based algorithm (msprime; Baumdicker et al 2022). The main advantage of slendr is that the whole simulative experiment can be performed entirely in the R environment, taking advantage of the many libraries available for geospatial and genomic data analysis, statistics, and visualisation. The open-source nature of this package, whose main aim is to make complex population genomics modelling more accessible, and the vibrant community of SLiM and tskit users will very likely make slendr widely used amongst the molecular ecology and evolutionary biology communities. Slendr handles real Earth cartographic data where users can design realistic demographic processes which characterise natural populations (i.e., expansions, displacement of large populations, interactions among populations, migrations, population splits, etc.) by changing spatial population boundaries across time and space. All in all, slendr is a very flexible and scalable framework to test the accuracy of spatial models, hypotheses about demography and selection, and interactions between organisms across space and time. REFERENCES Baumdicker, F., Bisschop, G., Goldstein, D., Gower, G., Ragsdale, A. P., Tsambos, G., ... & Kelleher, J. (2022). Efficient ancestry and mutation simulation with msprime 1.0. Genetics, 220(3), iyab229. https://doi.org/10.1093/genetics/iyab229 Currat, M., Ray, N., & Excoffier, L. (2004). SPLATCHE: a program to simulate genetic diversity taking into account environmental heterogeneity. Molecular Ecology Notes, 4(1), 139-142. https://doi.org/10.1046/j.1471-8286.2003.00582.x Haller, B. C., & Messer, P. W. (2017). SLiM 2: flexible, interactive forward genetic simulations. Molecular biology and evolution, 34(1), 230-240. https://doi.org/10.1093/molbev/msw211 Hoban, S., Bertorelle, G., & Gaggiotti, O. E. (2012). Computer simulations: tools for population and evolutionary genetics. Nature Reviews Genetics, 13(2), 110-122. https://doi.org/10.1038/nrg3130 Kelleher, J., Thornton, K. R., Ashander, J., & Ralph, P. L. (2018). Efficient pedigree recording for fast population genetics simulation. PLoS computational biology, 14(11), e1006581. https://doi.org/10.1371/journal.pcbi.1006581 Petr, M., Haller, B. C., Ralph, P. L., & Racimo, F. (2023). slendr: a framework for spatio-temporal population genomic simulations on geographic landscapes. bioRxiv, 2022.03.20.485041, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.03.20.485041 | slendr: a framework for spatio-temporal population genomic simulations on geographic landscapes | Martin Petr, Benjamin C. Haller, Peter L. Ralph, Fernando Racimo | <p style="text-align: justify;">One of the goals of population genetics is to understand how evolutionary forces shape patterns of genetic variation over time. However, because populations evolve across both time and space, most evolutionary proce... | | Bioinformatics & Computational Biology, Evolutionary Theory, Phylogeography & Biogeography, Population Genetics / Genomics | Emiliano Trucchi | 2022-09-14 12:57:56 |




