
Latest recommendations

| Id | Title * ▲ | Authors * | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
20 Sep 2017

An interaction between cancer progression and social environment in DrosophilaCancer and loneliness in DrosophilaRecommended by Ana Rivero based on reviews by Ana Rivero and Silvie HuijbenDrosophila flies may not be perceived as a quintessentially social animal, particularly when compared to their eusocial hymenopteran cousins. Although they have no parental care, division of labour or subfertile caste, fruit flies nevertheless exhibit an array of social phenotypes that are potentially comparable to those of their highly social relatives. In the wild, Drosophila adults cluster around food resources where courtship, mating activity and oviposition occur. Recent work has shown not only that social interactions in these clusters condition many aspects of the behaviour and physiology of the flies [1] but also, and perhaps more unexpectedly, that social isolation has a negative impact on their fitness [2]. Many studies in humans point to the role of social isolation as a source of stress that can induce and accelerate disease progression. The ultimate proof of the connection between social interaction and disease is however mired in confounding variables and alternative explanations so the subject, though crucial, remains controversial. With a series of elegant experiments using Drosophila flies that develop an inducible form of intestinal cancer, Dawson et al [3] show that cancer progresses more rapidly in flies maintained in isolation than in flies maintained with other cancerous flies. Further, cancerous flies kept with non-cancerous flies, fare just as badly as when kept alone. Their experiments suggest that this is due to the combined effect of healthy flies avoiding contact with cancerous flies (even though this is a non-contagious disease), and of cancerous flies having higher quality interactions with other cancerous flies than with healthy ones. Perceived isolation is therefore as pernicious as real isolation when it comes to cancer progression in these flies. Like all good research, this study opens up as many questions as it answers, in particular the why and wherefores of the flies’ extraordinary social behaviour in the face of disease. References [1] Camiletti AL and Thompson GJ. 2016. Drosophila as a genetically tractable model for social insect behavior. Frontiers in Ecology and Evolution, 4: 40. doi: 10.3389/fevo.2016.00040 [2] Ruan H and Wu C-F. 2008. Social interaction-mediated lifespan extension of Drosophila Cu/Zn superoxide dismutase mutants. Proceedings of the National Academy of Sciences, USA, 105: 7506-7510. doi: 10.1073/pnas.0711127105 [3] Dawson E, Bailly T, Dos Santos J, Moreno C, Devilliers M, Maroni B, Sueur C, Casali A, Ujvari B, Thomas F, Montagne J, Mery F. 2017. An interaction between cancer progression and social environment in Drosophila. BiorXiv, 143560, ver. 3 of 19th September 2017. doi: 10.1101/143560 | An interaction between cancer progression and social environment in Drosophila | Erika H. Dawson, Tiphaine P.M. Bailly, Julie Dos Santos , Céline Moreno, Maëlle Devilliers, Brigitte Maroni, Cédric Sueur, Andreu Casali, Beata Ujvari, Frederic Thomas, Jacques Montagne, Frederic Mery | The ecological benefits of sociality in gregarious species are widely acknowledged. However, only limited data is available on how the social environment influences non-communicable disease outcomes. For instance, despite extensive research over t... | | Behavior & Social Evolution, Evolutionary Ecology, Phenotypic Plasticity | Ana Rivero | 2017-05-30 08:55:16 | ||
23 Jan 2020

A novel workflow to improve multi-locus genotyping of wildlife species: an experimental set-up with a known model systemImproving the reliability of genotyping of multigene families in non-model organismsRecommended by François Rousset based on reviews by Sebastian Ernesto Ramos-Onsins, Helena Westerdahl and Thomas BigotThe reliability of published scientific papers has been the topic of much recent discussion, notably in the biomedical sciences [1]. Although small sample size is regularly pointed as one of the culprits, big data can also be a concern. The advent of high-throughput sequencing, and the processing of sequence data by opaque bioinformatics workflows, mean that sequences with often high error rates are produced, and that exact but slow analyses are not feasible. References [1] Ioannidis, J. P. A, Greenland, S., Hlatky, M. A., Khoury, M. J., Macleod, M. R., Moher, D., Schulz, K. F. and Tibshirani, R. (2014) Increasing value and reducing waste in research design, conduct, and analysis. The Lancet, 383, 166-175. doi: 10.1016/S0140-6736(13)62227-8 | A novel workflow to improve multi-locus genotyping of wildlife species: an experimental set-up with a known model system | Gillingham, Mark A. F., Montero, B. Karina, Wilhelm, Kerstin, Grudzus, Kara, Sommer, Simone and Santos, Pablo S. C. | <p>Genotyping novel complex multigene systems is particularly challenging in non-model organisms. Target primers frequently amplify simultaneously multiple loci leading to high PCR and sequencing artefacts such as chimeras and allele amplification... | | Bioinformatics & Computational Biology, Evolutionary Ecology, Genome Evolution, Molecular Evolution | François Rousset | Helena Westerdahl, Sebastian Ernesto Ramos-Onsins, Paul J. McMurdie , Arnaud Estoup, Vincent Segura, Jacek Radwan , Torbjørn Rognes , William Stutz , Kevin Vanneste , Thomas Bigot, Jill A. Hollenbach , Wieslaw Babik , Marie-Christin... | 2019-05-15 17:30:44 | |
15 Feb 2019

Architectural traits constrain the evolution of unisexual flowers and sexual segregation within inflorescences: an interspecific approachSometimes, sex is in the headRecommended by Juan Arroyo based on reviews by 3 anonymous reviewers based on reviews by 3 anonymous reviewers
Plants display an amazing diversity of reproductive strategies with and without sex. This diversity is particularly remarkable in flowering plants, as highlighted by Charles Darwin, who wrote several botanical books scrutinizing plant reproduction. One particularly influential work concerned floral variation [1]. Darwin recognized that flowers may present different forms within a single population, with or without sex specialization. The number of species concerned is small, but they display recurrent patterns, which made it possible for Darwin to invoke natural and sexual selection to explain them. Most of early evolutionary theory on the evolution of reproductive strategies was developed in the first half of the 20th century and was based on animals. However, the pioneering work by David Lloyd from the 1970s onwards excited interest in the diversity of plant sexual strategies as models for testing adaptive hypotheses and predicting reproductive outcomes [2]. The sex specialization of individual flowers and plants has since become one of the favorite topics of evolutionary biologists. However, attention has focused mostly on cases related to sex differentiation (dioecy and associated conditions [3]). Separate unisexual flower types on the same plant (monoecy and related cases, rendering the plant functionally hermaphroditic) have been much less studied, apart from their possible role in the evolution of dioecy [4] or their association with particular modes of pollination [5]. References [1] Darwin, C. (1877). The different forms of flowers on plants of the same species. John Murray. | Architectural traits constrain the evolution of unisexual flowers and sexual segregation within inflorescences: an interspecific approach | Rubén Torices, Ana Afonso, Arne A. Anderberg, José M. Gómez and Marcos Méndez | <p>Male and female unisexual flowers have repeatedly evolved from the ancestral bisexual flowers in different lineages of flowering plants. This sex specialization in different flowers often occurs within inflorescences. We hypothesize that inflor... | | Evolutionary Ecology, Morphological Evolution, Phenotypic Plasticity, Reproduction and Sex, Sexual Selection | Juan Arroyo | Jana Vamosi, Marcial Escudero, Anonymous | 2018-06-27 10:49:52 | |
21 Nov 2022

Artisanal and farmers bread making practices differently shape fungal species community composition in French sourdoughsThe variety of bread-making practices promotes diversity conservation in food microbial communitiesRecommended by Tatiana Giraud and Jeanne Ropars based on reviews by 2 anonymous reviewers
Domesticated organisms are excellent models for understanding ecology and evolution and they are important for our food production and safety. While less studied than plants and animals, micro-organisms have also been domesticated, in particular for food fermentation [1]. The most studied domesticated micro-organism is the yeast used to make wine, beer and bread, Saccharomyces cerevisiae [2, 3, 4]. Filamentous fungi used for cheese-making have recently gained interest, for example Penicillium roqueforti used to make blue cheeses and P. camemberti to make soft cheeses [5, 6, 7, 8]. As for plants and animals, domestication has led to beneficial traits for food production in fermenting fungi, but also to bottlenecks and degeneration [6, 7, 9]; P. camemberti for example does not produce enough spores any more for optimal culture and inoculation and P. roqueforti has lost sexual fertility [9]. The loss of genetic diversity and of species diversity in our food production system is concerning for multiple reasons : i) it jeopardizes future improvement in the face of global changes ; ii) it causes the loss of evolved diversity during centuries under human selection, and therefore of beneficial characteristics and specificities that we may never be able to recover ; iii) it leads to degeneration in the few cultivated strains; iv) it impoverishes the diversity of our food products and local adaptation of production practices. The study of domesticated fungi used for food fermentation has focused so far on the evolution of lineages and on their metabolic specificities. Microbiological assemblages and species diversity have been much less studied, while they likely also have a strong impact on the quality and safety of final products. This study by Elisa Michel and colleagues [10] addresses this question, using an interdisciplinary participatory research approach including bakers, psycho-sociologists and microbiologists to analyse bread-making practices and their impact on microbial communities in sourdough. Elisa Michel and colleagues [10] identified two distinct groups of bread-making practices based on interviews and surveys, with farmer-like practices (low bread production, use of ancient wheat populations, manual kneading, working at ambient temperature, long fermentation periods and no use of commercial baker’s yeast) versus more intensive, artisanal-like practices. Metabarcoding and microbial culture-based analyses showed that the well-known baker’s yeast, Saccharomyces cerevisiae, was, surprisingly, not the most common species in French organic sourdoughs. Kazachstania was the most represented yeast genus over all sourdoughs, both in terms of read abundance and of species diversity. Kazachstania species were also often dominant in individual sourdoughs, but Saccharomyces uvarum or Torulaspora delbrueckii could also be the dominant yeast species. Metabarcoding analyses further revealed that the composition of the fungal communities differed between the farmer-like and more intensive practices, representing the first evidence of the influence of artisanal practices on microbial communities. The fungal communities were impacted by a combination of bread-making variables including the type of wheat varieties, the length of fermentation, the quantity of bread made per week and the use of commercial yeast. Maintaining on farm less intensive bread-making practices, may allow the preservation of typical species and phenotypic diversity in microbial communities in sourdough. Farmer-like practices did not lead to higher diversity within sourdoughs but, overall, the diversity of bread-making practices allow maintaining a larger diversity in sourdoughs. For example, different Kazachstania species were most abundant in sourdoughs from artisanal-like and farmer-like practices. Interviews with the bakers suggested the role of dispersal of Kazachstania species in shaping sourdough microbial communities, dispersal occurring by seed exchanges, sourdough mixing or gifts, bread-making training in common or working in one another’s bakery. Nikolai Vavilov [11] had already highlighted for crops the importance of isolated cultures and selection in different farms for generating and preserving crop diversity, but also the importance of seed exchange for fostering adaptation. Furthermore, one of the yeast frequently found in artisanal sourdoughs, Kazachstania humilis, displayed phenotypic differences between sourdough and non-sourdough strains, suggesting domestication. The sourdough strains exhibited significantly higher CO2 production rate and a lower fermentation latency-phase time. The study by Elisa Michel and colleagues [10] is thus novel and inspiring in showing the importance of interdisciplinary studies, combining metabarcoding, microbiology and interviews for assessing the composition and diversity of microbial communities in human-made food, and in revealing the impact of artisanal-like bread-making practices in preserving microbial community diversity. Interdisciplinary studies are still rare but have already shown the importance of combining ethno-ecology, biology and evolution to decipher the role of human practices on genetic diversity in crops, animals and food microorganisms and to help preserving genetic resources [12]. For example, in the case of the bread wheat Triticum aestivum, such interdisciplinary studies have shown that genetic diversity has been shaped by farmers’ seed diffusion and farming practices [13]. We need more of such interdisciplinary studies on the impact of farmer versus industrial agricultural and food-making practices as we urgently need to preserve the diversity of micro-organisms used in food production that we are losing at a rapid pace [6, 7, 14]. References [1] Dupont J, Dequin S, Giraud T, Le Tacon F, Marsit S, Ropars J, Richard F, Selosse M-A (2017) Fungi as a Source of Food. Microbiology Spectrum, 5, 5.3.09. https://doi.org/10.1128/microbiolspec.FUNK-0030-2016 [2] Legras J-L, Galeote V, Bigey F, Camarasa C, Marsit S, Nidelet T, Sanchez I, Couloux A, Guy J, Franco-Duarte R, Marcet-Houben M, Gabaldon T, Schuller D, Sampaio JP, Dequin S (2018) Adaptation of S. cerevisiae to Fermented Food Environments Reveals Remarkable Genome Plasticity and the Footprints of Domestication. Molecular Biology and Evolution, 35, 1712–1727. https://doi.org/10.1093/molbev/msy066 [3] Bai F-Y, Han D-Y, Duan S-F, Wang Q-M (2022) The Ecology and Evolution of the Baker’s Yeast Saccharomyces cerevisiae. Genes, 13, 230. https://doi.org/10.3390/genes13020230 [4] Fay JC, Benavides JA (2005) Evidence for Domesticated and Wild Populations of Saccharomyces cerevisiae. PLOS Genetics, 1, e5. https://doi.org/10.1371/journal.pgen.0010005 [5] Ropars J, Rodríguez de la Vega RC, López-Villavicencio M, Gouzy J, Sallet E, Dumas É, Lacoste S, Debuchy R, Dupont J, Branca A, Giraud T (2015) Adaptive Horizontal Gene Transfers between Multiple Cheese-Associated Fungi. Current Biology, 25, 2562–2569. https://doi.org/10.1016/j.cub.2015.08.025 [6] Dumas E, Feurtey A, Rodríguez de la Vega RC, Le Prieur S, Snirc A, Coton M, Thierry A, Coton E, Le Piver M, Roueyre D, Ropars J, Branca A, Giraud T (2020) Independent domestication events in the blue-cheese fungus Penicillium roqueforti. Molecular Ecology, 29, 2639–2660. https://doi.org/10.1111/mec.15359 [7] Ropars J, Didiot E, Rodríguez de la Vega RC, Bennetot B, Coton M, Poirier E, Coton E, Snirc A, Le Prieur S, Giraud T (2020) Domestication of the Emblematic White Cheese-Making Fungus Penicillium camemberti and Its Diversification into Two Varieties. Current Biology, 30, 4441-4453.e4. https://doi.org/10.1016/j.cub.2020.08.082 [8] Caron T, Piver ML, Péron A-C, Lieben P, Lavigne R, Brunel S, Roueyre D, Place M, Bonnarme P, Giraud T, Branca A, Landaud S, Chassard C (2021) Strong effect of Penicillium roqueforti populations on volatile and metabolic compounds responsible for aromas, flavor and texture in blue cheeses. International Journal of Food Microbiology, 354, 109174. https://doi.org/10.1016/j.ijfoodmicro.2021.109174 [9] Ropars J, Lo Y-C, Dumas E, Snirc A, Begerow D, Rollnik T, Lacoste S, Dupont J, Giraud T, López-Villavicencio M (2016) Fertility depression among cheese-making Penicillium roqueforti strains suggests degeneration during domestication. Evolution, 70, 2099–2109. https://doi.org/10.1111/evo.13015 [10] Michel E, Masson E, Bubbendorf S, Lapicque L, Nidelet T, Segond D, Guézenec S, Marlin T, Devillers H, Rué O, Onno B, Legrand J, Sicard D, Bakers TP (2022) Artisanal and farmer bread making practices differently shape fungal species community composition in French sourdoughs. bioRxiv, 679472, ver. 6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/679472 [11] Vavilov NI, Vavylov MI, Dorofeev VF (1992) Origin and Geography of Cultivated Plants. Cambridge University Press. [12] Saslis-Lagoudakis CH, Clarke AC (2013) Ethnobiology: the missing link in ecology and evolution. Trends in Ecology & Evolution, 28, 67–68. https://doi.org/10.1016/j.tree.2012.10.017 [13] Thomas M, Demeulenaere E, Dawson JC, Khan AR, Galic N, Jouanne-Pin S, Remoue C, Bonneuil C, Goldringer I (2012) On-farm dynamic management of genetic diversity: the impact of seed diffusions and seed saving practices on a population-variety of bread wheat. Evolutionary Applications, 5, 779–795. https://doi.org/10.1111/j.1752-4571.2012.00257.x [14] Demeulenaere É, Lagrola M (2021) Des indicateurs pour accompagner “ les éleveurs de microbes” : Une communauté épistémique face au problème des laits “ paucimicrobiens ” dans la production fromagère au lait cru (1995-2015). Revue d’anthropologie des connaissances, 15. http://journals.openedition.org/rac/24953 | Artisanal and farmers bread making practices differently shape fungal species community composition in French sourdoughs | Elisa Michel, Estelle Masson, Sandrine Bubbendorf, Leocadie Lapicque, Thibault Nidelet, Diego Segond, Stephane Guezenec, Therese Marlin, Hugo deVillers, Olivier Rue, Bernard Onno, Judith Legrand, Delphine Sicard | <p style="text-align: justify;">Preserving microbial diversity in food systems is one of the many challenges to be met to achieve food security and quality. Although industrialization led to the selection and spread of specific fermenting microbia... | | Adaptation, Evolutionary Applications, Evolutionary Ecology | Tatiana Giraud | 2022-01-27 14:53:08 | ||
18 Jan 2017

POSTPRINT
Associative Mechanisms Allow for Social Learning and Cultural Transmission of String Pulling in an InsectCulture in BumblebeesRecommended by Caroline Nieberding and Jacques J. M. van AlphenThis is an original paper [1] addressing the question whether cultural transmission occurs in insects and studying the mechanisms of such transmission. Often, culture-like phenomena require relatively sophisticated learning mechanisms, for example imitation and/or teaching. In insects, seemingly complex processes of social information acquisition, can sometimes instead be mediated by relatively simple learning mechanisms suggesting that cultural processes may not necessarily require sophisticated learning abilities. An important quality of this paper is to describe neatly the experimental protocols used for such typically complex behavioural analyses, providing a detailed understanding of the results while it remains a joy to read. This becomes rare in high impact journals. In a clever experimental design, individual bumblebees are trained to pull an artificial flower from under a Plexiglas table to get access to a reward, by pulling a string attached to the flower. Individuals that have learnt this task are then shown to inexperienced bees while performing this task. This results in a large proportion of the inexperienced observers learning to pull the string and getting access to the reward. Finally, the authors could then document the spread of the string pulling skill amongst other workers in the colony. Even when the originally trained individuals had died, the skill of string-pulling persisted in the colony, as long as they were challenged with the task. This shows that cultural transmission takes place within a colony. The authors provide evidence that the transmission of this behavior among individuals relies on a mix of social learning by local enhancement (bees were attracted to the location where they had observed a demonstrator) and of non-social, individual learning (pulling the string is learned by trial and errors and not by direct imitation of the conspecific). Data also show that simple associative mechanisms are enough and that stimulus enhancement was involved (bees were attracted to the string when its location was concordant with that during prior observation). The cleverly designed experiments use a paradigm (string-pulling) which has often been used to investigate cognitive abilities in vertebrates. Comparison with such studies indicate that bees, in some aspects of their learning, may not be different from birds, dogs, or apes as they also relied on the perceptual feedback provided by their actions, resulting in target movement to learn string pulling. The results of the study suggest that the combination of relatively simple forms of social learning and trial-and-error learning can mediate the acquisition of new skills and that bumblebees possess the essential cognitive elements for cultural transmission and in a broader sense, that the capacity of culture may be present within most animals. Can we expect behavioural innovation such as string pulling to occur in nature? Bombus terrestris colonies can reach a total of several hundreds foragers. In the experiments, foragers needed on average 5 rounds of observations with different demonstrators to learn how to pull the string. As individuals forage in a meadow full of flowers and conspecifics, transmission of behavioural innovations by repeated observations shouldn’t strike us as something impossible. Would the behavior survive through the winter? Bumblebee colonies are seasonal in northern areas and in the Mediterranean area but tropical species persists for several years. In seasonal species, all the workers die before winter and only new queens overwinter. So there is no possibility for seasonal foragers to transmit the technique overwinter. Only queens could potentially transmit it to new foragers in spring. However flowers are different in autumn and spring. Therefore, what queens have learnt about flowers in autumn would unlikely be useful in spring (providing that they can remember it). However there is no reason why the technique couldn't be transmitted from a colony to another between spring to autumn. Such transmission of new behaviour would more easily persist in perennial social insect colonies, like honeybees. Importantly, the bees used in these experiments came from a company whose rearing conditions are unknown, and only a few colonies were used for each experiment. As learning ability has a genetic basis [2-3], colonies differ in their ability to learn [4]. In this regard, the authors showed variation between individual bumblebees and between bumblebee colonies in learning ability. Hence, we would wish to know more about the level of genetic diversity in the wild, and of genetic differentiation between tested colonies (were they independent replicates?), to extrapolate the results to what may happen in the wild. Excitingly, the authors found 2 true innovators among the >400 individuals that were tested at least once for 5 min who would solve such a task without stepwise training or observation of skilled demonstrators, showing that behavioural innovation can occur in very small numbers of individuals, provided that an ecological trigger is provided (food reward). Hence this study shows that all ingredients for the long proposed “social heredity” theory proposed by Baldwin in 1896 are available in this organism, suggesting that social transmission of behavioural innovations could technically act as an additional mechanism for adaptive evolution [5], next to genetic evolution that may take longer to produce adaptive evolution. The question remains whether the behavioural innovations are arising from standing genetic variation in the bees, or do not need a firm genetic background to appear. References [1] Alem S, Perry CJ, Zhu X, Loukola OJ, Ingraham T, Søvik E, Chittka L. 2016. Associative mechanisms allow for social learning and cultural transmission of string pulling in an insect. PloS Biology 14:e1002564. doi: 10.1371/journal.pbio.1002564 [2] Mery F, Kawecki TJ. 2002. Experimental evolution of learning ability in fruit flies. Proceeding of the National Academy of Science USA 99:14274-14279. doi: 10.1073/pnas.222371199 [3] Mery F, Belay AT, So AKC, Sokolowski MB, Kawecki TJ. 2007. Natural polymorphism affecting learning and memory in Drosophila. Proceeding of the National Academy of Science USA 104:13051-13055. doi: 10.1073/pnas.0702923104 [4] Raine NE, Chittka L. 2008. The correlation of learning speed and natural foraging success in bumble-bees. Proceeding of the Royal Society of London 275: 803-808. doi : 10.1098/rspb.2007.1652 [5] Baldwin JM. 1896. A New Factor in Evolution. American Naturalist 30:441-451 and 536-553. doi: 10.1086/276408 | Associative Mechanisms Allow for Social Learning and Cultural Transmission of String Pulling in an Insect | Alem S, Perry CJ, Zhu X, Loukola OJ, Ingraham T, Søvik E, Chittka L | Social insects make elaborate use of simple mechanisms to achieve seemingly complex behavior and may thus provide a unique resource to discover the basic cognitive elements required for culture, i.e., group-specific behaviors that spread from “inn... | | Behavior & Social Evolution, Evolutionary Ecology, Non Genetic Inheritance, Phenotypic Plasticity | Caroline Nieberding | 2017-01-18 10:49:03 | ||
12 Jul 2017

Assortment of flowering time and defense alleles in natural Arabidopsis thaliana populations suggests co-evolution between defense and vegetative lifespan strategiesTowards an integrated scenario to understand evolutionary patterns in A. thalianaRecommended by Xavier Picó based on reviews by Rafa Rubio de Casas and Xavier PicóNobody can ignore that a full understanding of evolution requires an integrated approach from both conceptual and methodological viewpoints. Although some life-history traits, e.g. flowering time, have long been receiving more attention than others, in many cases because the former are more workable than the latter, we must acknowledge that our comprehension about how evolution works is strongly biased and limited. In the Arabidopsis community, such an integration is making good progress as an increasing number of research groups worldwide are changing the way in which evolution is put to the test. This manuscript [1] is a good example of that as the authors raise an important issue in evolutionary biology by combining gene expression and flowering time data from different sources. In particular, the authors explore how variation in flowering time, which determines lifespan, and host immunity defenses co-vary, which is interpreted in terms of co-evolution between the two traits. Interestingly, the authors go beyond that pattern by separating lifespan-dependent from lifespan–independent defense genes, and by showing that defense genes with variants known to impact fitness in the field are among the genes whose expression co-varies most strongly with flowering time. Finally, these results are supported by a simple mathematical model indicating that such a relationship can also be expected theoretically. Overall, the readers will find many conceptual and methodological elements of interest in this manuscript. The idea that evolution is better understood under the scope of life history variation is really exciting and challenging, and in my opinion on the right track for disentangling the inherent complexities of evolutionary research. However, only when we face complexity, we also face its costs and burdens. In this particular case, the well-known co-variation between seed dormancy and flowering time is a missing piece, as well as the identification of (variation in) putative selective pressures accounting for the co-evolution between defense mechanisms and life history (seed dormancy vs. flowering time) along environmental gradients. More intellectual, technical and methodological challenges that with no doubt are totally worth it. Reference [1] Glander S, He F, Schmitz G, Witten A, Telschow A, de Meaux J. 2017. Assortment of flowering time and defense alleles in natural Arabidopsis thaliana populations suggests co-evolution between defense and vegetative lifespan strategies. bioRxiv ver.1 of June 19, 2017. doi: 10.1101/131136 | Assortment of flowering time and defense alleles in natural Arabidopsis thaliana populations suggests co-evolution between defense and vegetative lifespan strategies | Glander S, He F, Schmitz G, Witten A, Telschow A, de Meaux J | The selective impact of pathogen epidemics on host defenses can be strong but remains transient. By contrast, life-history shifts can durably and continuously modify the balance between costs and benefits of immunity, which arbitrates the evolutio... | | Adaptation, Evolutionary Ecology, Expression Studies, Life History, Phenotypic Plasticity, Quantitative Genetics, Species interactions | Xavier Picó | Sophie Karrenberg, Rafa Rubio de Casas, Xavier Picó | 2017-06-21 10:57:14 | |
13 Dec 2016

POSTPRINT
A supergene determines highly divergent male reproductive morphs in the ruffSupergene Control of a Reproductive PolymorphismRecommended by Thomas Flatt and Laurent KellerTwo back-to-back papers published earlier this year in Nature Genetics provide compelling evidence for the control of a male reproductive polymorphism in a wading bird by a "supergene", a cluster of tightly linked genes [1-2]. The bird in question, the ruff (Philomachus pugnax), has a rather unusual reproductive system that consists of three distinct types of males ("reproductive morphs"): aggressive "independents" who represent the majority of males; a smaller fraction of non-territorial "satellites" who are submissive towards "independents"; and "faeders" who mimic females and are rare. Previous work has shown that the male morphs differ in major aspects of mating and aggression behavior, plumage coloration and body size, and that – intriguingly – this complex multi-trait polymorphism is apparently controlled by a single autosomal Mendelian locus with three alleles [3]. To uncover the genetic control of this polymorphism two independent teams, led by Terry Burke [1] and Leif Andersson [2], have set out to analyze the genomes of male ruffs. Using a combination of genomics and genetics, both groups managed to pin down the supergene locus and map it to a non-recombining, 4.5 Mb large inversion which arose 3.8 million years ago. While "independents" are homozygous for the ancestral uninverted sequence, "satellites" and "faeders" carry evolutionarily divergent, dominant alternative haplotypes of the inversion. Thus, as in several other notable cases, for example the supergene control of disassortative mating, aggressiveness and plumage color in white-throated sparrows [4], of mimicry in Heliconius and Papilio butterflies [5-6], or of social structure in ants [7], an inversion – behaving as a single "locus" – underpins the mechanistic basis of the supergene. More generally, and beyond inversions, a growing number of studies now shows that selection can favor the evolution of suppressed recombination, thereby leading to the emergence of clusters of tightly linked loci which can then control – presumably due to polygenic gene action – a suite of complex phenotypes [8-10]. A largely unresolved question in this field concerns the identity of the causative alleles and loci within a given supergene. Recent progress on this question has been made for example in Papilio polytes butterflies where a mimicry supergene has been found to involve – surprisingly – only a single but large gene: multiple mimicry alleles in the doublesex gene are maintained in strong linkage disequilibrium via an inversion. It will clearly be of great interest to see future examples of such a fine-scale genetic dissection of supergenes. In conclusion, we were impressed by the data and analyses of Küpper et al. [1] and Lamichhaney et al. [2]: both papers beautifully illustrate how genomics and evolutionary ecology can be combined to make new, exciting discoveries. Both papers will appeal to readers with an interest in supergenes, inversions, the interplay of selection and recombination, or the genetic control of complex phenotypes. References [1] Küpper C, Stocks M, Risse JE, dos Remedios N, Farrell LL, McRae SB, Morgan TC, Karlionova N, Pinchuk P, Verkuil YI, et al. 2016. A supergene determines highly divergent male reproductive morphs in the ruff. Nature Genetics 48:79-83. doi: 10.1038/ng.3443 [2] Lamichhaney S, Fan G, Widemo F, Gunnarsson U, Thalmann DS, Hoeppner MP, Kerje S, Gustafson U, Shi C, Zhang H, et al. 2016. Structural genomic changes underlie alternative reproductive strategies in the ruff (Philomachus pugnax). Nature Genetics 48:84-88. doi: 10.1038/ng.3430 [3] Lank DB, Smith CM, Hanotte O, Burke T, Cooke F. 1995. Genetic polymorphism for alternative mating behaviour in lekking male ruff Philomachus pugnax. Nature 378:59-62. doi: 10.1038/378059a0 [4] Tuttle Elaina M, Bergland Alan O, Korody Marisa L, Brewer Michael S, Newhouse Daniel J, Minx P, Stager M, Betuel A, Cheviron Zachary A, Warren Wesley C, et al. 2016. Divergence and Functional Degradation of a Sex Chromosome-like Supergene. Current Biology 26:344-350. doi: 10.1016/j.cub.2015.11.069 [5] Joron M, Frezal L, Jones RT, Chamberlain NL, Lee SF, Haag CR, Whibley A, Becuwe M, Baxter SW, Ferguson L, et al. 2011. Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 477:203-206. doi: 10.1038/nature10341 [6] Kunte K, Zhang W, Tenger-Trolander A, Palmer DH, Martin A, Reed RD, Mullen SP, Kronforst MR. 2014. doublesex is a mimicry supergene. Nature 507:229-232. doi: 10.1038/nature13112 [7] Wang J, Wurm Y, Nipitwattanaphon M, Riba-Grognuz O, Huang Y-C, Shoemaker D, Keller L. 2013. A Y-like social chromosome causes alternative colony organization in fire ants. Nature 493:664-668. doi: 10.1038/nature11832 [8] Thompson MJ, Jiggins CD. 2014. Supergenes and their role in evolution. Heredity 113:1-8. doi: 10.1038/hdy.2014.20 [9] Schwander T, Libbrecht R, Keller L. 2014. Supergenes and Complex Phenotypes. Current Biology 24:R288-R294. doi: 10.1016/j.cub.2014.01.056 [10] Charlesworth D. 2015. The status of supergenes in the 21st century: recombination suppression in Batesian mimicry and sex chromosomes and other complex adaptations. Evolutionary Applications 9:74-90. doi: 10.1111/eva.12291 | A supergene determines highly divergent male reproductive morphs in the ruff | Küpper C, Stocks M, Risse JE, dos Remedios N, Farrell LL, McRae SB, Morgan TC, Karlionova N, Pinchuk P, Verkuil YI, et al. | Three strikingly different alternative male mating morphs (aggressive 'independents', semicooperative 'satellites' and female-mimic 'faeders') coexist as a balanced polymorphism in the ruff, *Philomachus pugnax*, a lek-breeding wading bird1, 2, 3.... | | Adaptation, Genotype-Phenotype, Life History, Population Genetics / Genomics, Reproduction and Sex | Thomas Flatt | 2016-12-13 17:28:13 | ||
20 Jan 2020

A young age of subspecific divergence in the desert locust Schistocerca gregaria, inferred by ABC Random ForestEstimating recent divergence history: making the most of microsatellite data and Approximate Bayesian Computation approachesRecommended by Takeshi Kawakami and Concetta Burgarella based on reviews by Michael D Greenfield and 2 anonymous reviewersThe present-day distribution of extant species is the result of the interplay between their past population demography (e.g., expansion, contraction, isolation, and migration) and adaptation to the environment. Shedding light on the timing and magnitude of key demographic events helps identify potential drivers of such events and interaction of those drivers, such as life history traits and past episodes of environmental shifts. The understanding of the key factors driving species evolution gives important insights into how the species may respond to changing conditions, which can be particularly relevant for the management of harmful species, such as agricultural pests (e.g. [1]). Meaningful demographic inferences present major challenges. These include formulating evolutionary scenarios fitting species biology and the eco-geographical context and choosing informative molecular markers and accurate quantitative approaches to statistically compare multiple demographic scenarios and estimate the parameters of interest. A further issue comes with result interpretation. Accurately dating the inferred events is far from straightforward since reliable calibration points are necessary to translate the molecular estimates of the evolutionary time into absolute time units (i.e. years). This can be attempted in different ways, such as by using fossil and archaeological records, heterochronous samples (e.g. ancient DNA), and/or mutation rate estimated from independent data (e.g. [2], [3] for review). Nonetheless, most experimental systems rarely meet these conditions, hindering the comprehensive interpretation of results. The contribution of Chapuis et al. [4] addresses these issues to investigate the recent history of the African insect pest Schistocerca gregaria (desert locust). They apply Approximate Bayesian Computation-Random Forest (ABC-RF) approaches to microsatellite markers. Owing to their fast mutation rate microsatellite markers offer at least two advantages: i) suitability for analyzing recently diverged populations, and ii) direct estimate of the germline mutation rate in pedigree samples. The work of Chapuis et al. [4] benefits of both these advantages, since they have estimates of mutation rate and allele size constraints derived from germline mutations in the species [5]. The main aim of the study is to infer the history of divergence of the two subspecies of the desert locust, which have spatially disjoint distribution corresponding to the dry regions of North and West-South Africa. They first use paleo-vegetation maps to formulate hypotheses about changes in species range since the last glacial maximum. Based on them, they generate 12 divergence models. For the selection of the demographic model and parameter estimation, they apply the recently developed ABC-RF approach, a powerful inferential tool that allows optimizing the use of summary statistics information content, among other advantages [6]. Some methodological novelties are also introduced in this work, such as the computation of the error associated with the posterior parameter estimates under the best scenario. The accuracy of timing estimate is assured in two ways: i) by the use of microsatellite markers with known evolutionary dynamics, as underlined above, and ii) by assessing the divergence time threshold above which posterior estimates are likely to be biased by size homoplasy and limits in allele size range [7]. The best-supported model suggests a recent divergence event of the subspecies of S. gregaria (around 2.6 kya) and a reduction of populations size in one of the subspecies (S. g. flaviventris) that colonized the southern distribution area. As such, results did not support the hypothesis that the southward colonization was driven by the expansion of African dry environments associated with the last glacial maximum, as it has been postulated for other arid-adapted species with similar African disjoint distributions [8]. The estimated time of divergence points at a much more recent origin for the two subspecies, during the late Holocene, in a period corresponding to fairly stable arid conditions similar to current ones [9,10]. Although the authors cannot exclude that their microsatellite data bear limited information on older colonization events than the last one, they bring arguments in favour of alternative explanations. The hypothesis privileged does not involve climatic drivers, but the particularly efficient dispersal behaviour of the species, whose individuals are able to fly over long distances (up to thousands of kilometers) under favourable windy conditions. A single long-distance dispersal event by a few individuals would explain the genetic signature of the bottleneck. There is a growing number of studies in phylogeography in arid regions in the Southern hemisphere, but the impact of past climate changes on the species distribution in this region remains understudied relative to the Northern hemisphere [11,12]. The study presented by Chapuis et al. [4] offers several important insights into demographic changes and the evolutionary history of an agriculturally important pest species in Africa, which could also mirror the history of other organisms in the continent. As the authors point out, there are necessarily some uncertainties associated with the models of past ecosystems and climate, especially for Africa. Interestingly, the authors argue that the information on paleo-vegetation turnover was more informative than climatic niche modeling for the purpose of their study since it made them consider a wider range of bio-geographical changes and in turn a wider range of evolutionary scenarios (see discussion in Supplementary Material). Microsatellite markers have been offering a useful tool in population genetics and phylogeography for decades, but their popularity is perhaps being taken over by single nucleotide polymorphism (SNP) genotyping and whole-genome sequencing (WGS) (the peak year of the number of the publication with “microsatellite” is in 2012 according to PubMed). This study reaffirms the usefulness of these classic molecular markers to estimate past demographic events, especially when species- and locus-specific microsatellite mutation features are available and a powerful inferential approach is adopted. Nonetheless, there are still hurdles to overcome, such as the limitations in scenario choice associated with the simulation software used (e.g. not allowing for continuous gene flow in this particular case), which calls for further improvement of simulation tools allowing for more flexible modeling of demographic events and mutation patterns. In sum, this work not only contributes to our understanding of the makeup of the African biodiversity but also offers a useful statistical framework, which can be applied to a wide array of species and molecular markers (microsatellites, SNPs, and WGS). References [1] Lehmann, P. et al. (2018). Complex responses of global insect pests to climate change. bioRxiv, 425488. doi: https://dx.doi.org/10.1101/425488 [2] Donoghue, P. C., & Benton, M. J. (2007). Rocks and clocks: calibrating the Tree of Life using fossils and molecules. Trends in Ecology & Evolution, 22(8), 424-431. doi: https://dx.doi.org/10.1016/j.tree.2007.05.005 [3] Ho, S. Y., Lanfear, R., Bromham, L., Phillips, M. J., Soubrier, J., Rodrigo, A. G., & Cooper, A. (2011). Time‐dependent rates of molecular evolution. Molecular ecology, 20(15), 3087-3101. doi: https://dx.doi.org/10.1111/j.1365-294X.2011.05178.x [4] Chapuis, M.-P., Raynal, L., Plantamp, C., Meynard, C. N., Blondin, L., Marin, J.-M. and Estoup, A. (2020). A young age of subspecific divergence in the desert locust Schistocerca gregaria, inferred by ABC Random Forest. bioRxiv, 671867, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: https://dx.doi.org/10.1101/671867 5] Chapuis, M.-P., Plantamp, C., Streiff, R., Blondin, L., & Piou, C. (2015). Microsatellite evolutionary rate and pattern in Schistocerca gregaria inferred from direct observation of germline mutations. Molecular ecology, 24(24), 6107-6119. doi: https://dx.doi.org/10.1111/mec.13465 [6] Raynal, L., Marin, J. M., Pudlo, P., Ribatet, M., Robert, C. P., & Estoup, A. (2018). ABC random forests for Bayesian parameter inference. Bioinformatics, 35(10), 1720-1728. doi: https://dx.doi.org/10.1093/bioinformatics/bty867 [7] Estoup, A., Jarne, P., & Cornuet, J. M. (2002). Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Molecular ecology, 11(9), 1591-1604. doi: https://dx.doi.org/10.1046/j.1365-294X.2002.01576.x [8] Moodley, Y. et al. (2018). Contrasting evolutionary history, anthropogenic declines and genetic contact in the northern and southern white rhinoceros (Ceratotherium simum). Proceedings of the Royal Society B, 285(1890), 20181567. doi: https://dx.doi.org/10.1098/rspb.2018.1567 [9] Kröpelin, S. et al. (2008). Climate-driven ecosystem succession in the Sahara: the past 6000 years. science, 320(5877), 765-768. doi: https://dx.doi.org/10.1126/science.1154913 [10] Maley, J. et al. (2018). Late Holocene forest contraction and fragmentation in central Africa. Quaternary Research, 89(1), 43-59. doi: https://dx.doi.org/10.1017/qua.2017.97 [11] Beheregaray, L. B. (2008). Twenty years of phylogeography: the state of the field and the challenges for the Southern Hemisphere. Molecular Ecology, 17(17), 3754-3774. doi: https://dx.doi.org/10.1111/j.1365-294X.2008.03857.x [12] Dubey, S., & Shine, R. (2012). Are reptile and amphibian species younger in the Northern Hemisphere than in the Southern Hemisphere?. Journal of evolutionary biology, 25(1), 220-226. doi: https://dx.doi.org/10.1111/j.1420-9101.2011.02417.x ***** A video about this preprint is available here: | A young age of subspecific divergence in the desert locust Schistocerca gregaria, inferred by ABC Random Forest | Marie-Pierre Chapuis, Louis Raynal, Christophe Plantamp, Christine N. Meynard, Laurence Blondin, Jean-Michel Marin, Arnaud Estoup | <p>Dating population divergence within species from molecular data and relating such dating to climatic and biogeographic changes is not trivial. Yet it can help formulating evolutionary hypotheses regarding local adaptation and future responses t... | | Bioinformatics & Computational Biology, Evolutionary Applications, Phylogeography & Biogeography, Population Genetics / Genomics | Takeshi Kawakami | 2019-06-20 10:31:15 | ||
30 Mar 2023
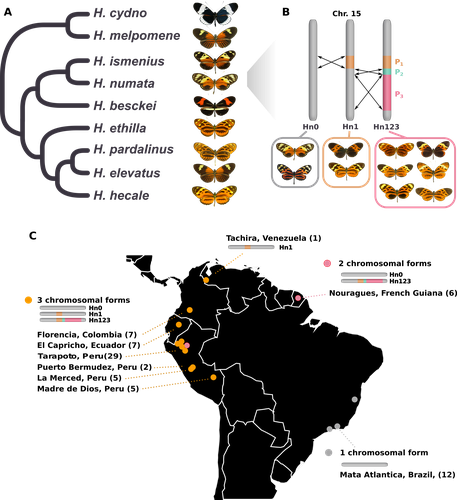
Balancing selection at a wing pattern locus is associated with major shifts in genome-wide patterns of diversity and gene flow in a butterflyIs genetic diversity enhanced by a supergene?Recommended by Chris Jiggins based on reviews by Christelle Fraïsse and 2 anonymous reviewersThe butterfly species Heliconius numata has a remarkable wing pattern polymorphism, with multiple pattern morphs all controlled by a single genetic locus, which harbours multiple inversions. Each morph is a near-perfect mimic of a species in the fairly distantly related genus of butterflies, Melinaea. The article by Rodríguez de Cara et al (2023) argues that the balanced polymorphism at this single wing patterning locus actually has a major effect on genetic diversity across the whole genome. First, polymorphic populations within H. numata are more dioverse than those without polymorphism. Second, H. numata is more genetically diverse than other related species and finally reconstruction of historical demography suggests that there has been a recent increase in effective population size, putatively associated with the acquisition of the supergene polymorphism. The supergene itself generates disassortative mating, such that morphs prefer to mate with others dissimilar to themselves - in this way it is similar to mechanisms for preventing inbreeding such as self-incompatibility loci in plants. This provides a potential mechanism whereby non-random mating patterns could increase effective population size. The authors also explore this mechanism using forward simulations, and show that mating patterns at a single locus can influence linked genetic diversity over a large scale. Overall, this is an intriguing study, which suggests a far more widespread genetic impact of a single locus than might be expected. There are interesting parallels with mechanisms of inbreeding prevention in plants, such as the Pin/Thrum polymorphism in Primula, which also rely on mating patterns determined by a single locus but presumably also influence genetic diversity genome-wide by promoting outbreeding. REFERENCES Rodríguez de Cara MÁ, Jay P, Rougemont Q, Chouteau M, Whibley A, Huber B, Piron-Prunier F, Ramos RR, Freitas AVL, Salazar C, Silva-Brandão KL, Torres TT, Joron M (2023) Balancing selection at a wing pattern locus is associated with major shifts in genome-wide patterns of diversity and gene flow. bioRxiv, 2021.09.29.462348, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.09.29.462348 | Balancing selection at a wing pattern locus is associated with major shifts in genome-wide patterns of diversity and gene flow in a butterfly | María Ángeles Rodríguez de Cara, Paul Jay, Quentin Rougemont, Mathieu Chouteau, Annabel Whibley, Barbara Huber, Florence Piron-Prunier, Renato Rogner Ramos, André V. L. Freitas, Camilo Salazar, Karina Lucas Silva-Brandão, Tatiana Texeira Torres, M... | <p style="text-align: justify;">Selection shapes genetic diversity around target mutations, yet little is known about how selection on specific loci affects the genetic trajectories of populations, including their genomewide patterns of diversity ... | | Evolutionary Ecology, Genome Evolution, Hybridization / Introgression, Population Genetics / Genomics | Chris Jiggins | 2021-10-13 17:54:33 | ||
15 Dec 2016

POSTPRINT
Basidiomycete yeasts in the cortex of ascomycete macrolichensNew partner at the core of macrolichen diversityRecommended by Enric Frago and Benoit FaconIt has long been known that most multicellular eukaryotes rely on microbial partners for a variety of functions including nutrition, immune reactions and defence against enemies. Lichens are probably the most popular example of a symbiosis involving a photosynthetic microorganism (an algae, a cyanobacteria or both) living embedded within the filaments of a fungus (usually an ascomycete). The latter is the backbone structure of the lichen, whereas the former provides photosynthetic products. Lichens are unique among symbioses because the structures the fungus and the photosynthetic microorganism form together do not resemble any of the two species living in isolation. Classic textbook examples like lichens are not often challenged and this is what Toby Spribille and his co-authors did with their paper published in July 2016 in Science [1]. This story started with the study of two species of macrolichens from the class of Lecanoromycetes that are commonly found in the mountains of Montana (US): Bryoria fremontii and B. tortuosa. For more than 90 years, these species have been known to differ in their chemical composition and colour, but studies performed so far failed in finding differences at the molecular level in both the mycobiont and the photobiont. These two species were therefore considered as nomenclatural synonyms, and the origin of their differences remained elusive. To solve this mystery, the authors of this work performed a transcriptome-wide analysis that, relative to previous studies, expanded the taxonomic range to all Fungi. This analysis revealed higher abundances of a previously unknown basidiomycete yeast from the genus Cyphobasidium in one of the lichen species, a pattern that was further confirmed by combining microscopy imaging and the fluorescent in situ hybridisation technique (FISH). Finding out that a previously unknown micro-organism changes the colour and the chemical composition of an organism is surprising but not new. For instance, bacterial symbionts are able to trigger colour changes in some insect species [2], and endophyte fungi are responsible for the production of defensive compounds in the leaves of several grasses [3]. The study by Spribille and his co-authors is fascinating because it demonstrates that Cyphobasidium yeasts have played a key role in the evolution and diversification of Lecanoromycetes, one of the most diverse classes of macrolichens. Indeed these basidiomycete yeasts were not only found in Bryoria but in 52 other lichen genera from all six continents, and these included 42 out of 56 genera in the family Parmeliaceae. Most of these sequences formed a highly supported monophyletic group, and a molecular clock revealed that the origin of many macrolichen groups occurred around the same time Cyphobasidium yeasts split from Cystobasidium, their nearest relatives. This newly discovered passenger is therefore an ancient inhabitant of lichens and has driven the evolution of this emblematic group of organisms. This study raises an important question on the stability of complex symbiotic partnerships. In intimate obligatory symbioses the evolutionary interests of both partners are often identical and what is good for one is also good for the other. This is the case of several insects that feed on poor diets like phloem and xylem sap, and which carry vertically-transmitted symbionts that provide essential nutrients. Molecular phylogenetic studies have repeatedly shown that in several insect groups transition to phloem or xylem feeding occurred at the same time these nutritional symbionts were acquired [4]. In lichens, an outstanding question is to know what was the key feature Cyphobasidium yeasts brought to the symbiosis. As suggested by the authors, these yeasts are likely to be involved in the production of secondary defensive metabolites and architectural structures, but, are these services enough to explain the diversity found in macrolichens? This paper is an appealing example of a multipartite symbiosis where the different partners share an ancient evolutionary history. References [1] Spribille T, Tuovinen V, Resl P, et al. 2016. Basidiomycete yeasts in the cortex of ascomycete macrolichens. Science 353:488–92. doi: 10.1126/science.aaf8287 [2] Tsuchida T, Koga R, Horikawa M, et al. 2010. Symbiotic Bacterium Modifies Aphid Body Color. Science 330:1102–1104. doi: 10.1126/science.1195463 [3] Clay K. 1988. Fungal Endophytes of Grasses: A Defensive Mutualism between Plants and Fungi. Ecology 69:10–16. doi: 10.2307/1943155 [4] Moran NA. 2007. Symbiosis as an adaptive process and source of phenotypic complexity. Proceeding of the National Academy of Science USA 104:8627–8633. doi: 10.1073/pnas.0611659104 | Basidiomycete yeasts in the cortex of ascomycete macrolichens | Spribille T, Tuovinen V, Resl P, et al. | For over 140 years, lichens have been regarded as a symbiosis between a single fungus, usually an ascomycete, and a photosynthesizing partner. Other fungi have long been known to occur as occasional parasites or endophytes, but the one lichen–one ... | | Adaptation, Evolutionary Ecology, Genome Evolution, Genotype-Phenotype, Life History, Macroevolution, Molecular Evolution, Phylogenetics / Phylogenomics, Speciation, Species interactions | Enric Frago | 2016-12-15 05:46:14 |




