
Latest recommendations

| Id | Title▲ | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
01 Jul 2022

Genomic evidence of paternal genome elimination in the globular springtail Allacma fuscaPressing NGS data through the mill of Kmer spectra and allelic coverage ratios in order to scan reproductive modes in non-model speciesRecommended by Nicolas Bierne based on reviews by Paul Simion and 2 anonymous reviewersThe genomic revolution has given us access to inexpensive genetic data for any species. Simultaneously we have lost the ability to easily identify chimerism in samples or some unusual deviations from standard Mendelian genetics. Methods have been developed to identify sex chromosomes, characterise the ploidy, or understand the exact form of parthenogenesis from genomic data. However, we rarely consider that the tissues we extract DNA from could be a mixture of cells with different genotypes or karyotypes. This can nonetheless happen for a variety of (fascinating) reasons such as somatic chromosome elimination, transmissible cancer, or parental genome elimination. Without a dedicated analysis, it is very easy to miss it. In this preprint, Jaron et al. (2022) used an ingenious analysis of whole individual NGS data to test the hypothesis of paternal genome elimination in the globular springtail Allacma fusca. The authors suspected that a high fraction of the whole body of males is made of sperm in this species and if this species undergoes paternal genome elimination, we would expect that sperm would only contain maternally inherited chromosomes. Given the reference genome was highly fragmented, they developed a two-tissue model to analyse Kmer spectra and obtained confirmation that around one-third of the tissue was sperm in males. This allowed them to test whether coverage patterns were consistent with the species exhibiting paternal genome elimination. They combined their estimation of the fraction of haploid tissue with allele coverages in autosomes and the X chromosome to obtain support for a bias toward one parental allele, suggesting that all sperm carries the same parental haplotype. It could be the maternal or the paternal alleles, but paternal genome elimination is most compatible with the known biology of Arthropods. SNP calling was used to confirm conclusions based on the analysis of the raw pileups. I found this study to be a good example of how a clever analysis of Kmer spectra and allele coverages can provide information about unusual modes of reproduction in a species, even though it does not have a well-assembled genome yet. As advocated by the authors, routine inspection of Kmer spectra and allelic read-count distributions should be included in the best practice of NGS data analysis. They provide the method to identify paternal genome elimination but also the way to develop similar methods to detect another kind of genetic chimerism in the avalanche of sequence data produced nowadays. References Jaron KS, Hodson CN, Ellers J, Baird SJ, Ross L (2022) Genomic evidence of paternal genome elimination in the globular springtail Allacma fusca. bioRxiv, 2021.11.12.468426, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.11.12.468426 | Genomic evidence of paternal genome elimination in the globular springtail Allacma fusca | Kamil S. Jaron, Christina N. Hodson, Jacintha Ellers, Stuart JE Baird, Laura Ross | <p style="text-align: justify;">Paternal genome elimination (PGE) - a type of reproduction in which males inherit but fail to pass on their father’s genome - evolved independently in six to eight arthropod clades. Thousands of species, including s... | | Genome Evolution, Reproduction and Sex | Nicolas Bierne | 2021-11-18 00:09:43 | ||
19 Feb 2018

Genomic imprinting mediates dosage compensation in a young plant XY systemDosage compensation by upregulation of maternal X alleles in both males and females in young plant sex chromosomesRecommended by Tatiana Giraud and Judith Mank based on reviews by 3 anonymous reviewersSex chromosomes evolve as recombination is suppressed between the X and Y chromosomes. The loss of recombination on the sex-limited chromosome (the Y in mammals) leads to degeneration of both gene expression and gene content for many genes [1]. Loss of gene expression or content from the Y chromosome leads to differences in gene dose between males and females for X-linked genes. Because expression levels are often correlated with gene dose [2], these hemizygous genes have a lower expression levels in the heterogametic sex. This in turn disrupts the stoichiometric balance among genes in protein complexes that have components on both the sex chromosomes and autosomes [3], which could have serious deleterious consequences for the heterogametic sex. References | Genomic imprinting mediates dosage compensation in a young plant XY system | Aline Muyle, Niklaus Zemp, Cecile Fruchard, Radim Cegan, Jan Vrana, Clothilde Deschamps, Raquel Tavares, Franck Picard, Roman Hobza, Alex Widmer, Gabriel Marais | <p>During the evolution of sex chromosomes, the Y degenerates and its expression gets reduced relative to the X and autosomes. Various dosage compensation mechanisms that recover ancestral expression levels in males have been described in animals.... | | Bioinformatics & Computational Biology, Expression Studies, Genome Evolution, Molecular Evolution, Reproduction and Sex | Tatiana Giraud | 2017-09-20 20:39:46 | ||
08 Jan 2024
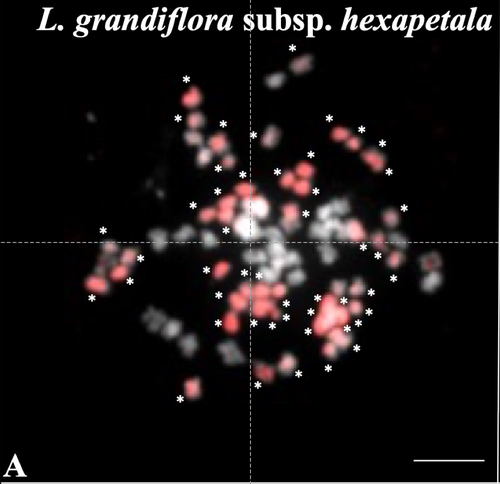
Genomic relationships among diploid and polyploid species of the genus Ludwigia L. section Jussiaea using a combination of molecular cytogenetic, morphological, and crossing investigationsDeciphering the genomic composition of tetraploid, hexaploid and decaploid Ludwigia L. species (section Jussiaea)Recommended by Malika AINOUCHE based on reviews by Alex BAUMEL and Karol MARHOLDPolyploidy, which results in the presence of more than two sets of homologous chromosomes represents a major feature of plant genomes that have undergone successive rounds of duplication followed by more or less rapid diploidization during their evolutionary history. Polyploid complexes containing diploid and derived polyploid taxa are excellent model systems for understanding the short-term consequences of whole genome duplication, and have been particularly well-explored in evolutionary ecology (Ramsey and Ramsey 2014, Rice et al. 2019). Many polyploids (especially when resulting from interspecific hybridization, i.e. allopolyploids) are successful invaders (te Beest et al. 2012) as a result of rapid genome dynamics, functional novelty, and trait evolution. The origin (parental legacy) and modes of formation of polyploids have a critical impact on the subsequent polyploid evolution. Thus, elucidation of the genomic composition of polyploids is fundamental to understanding trait evolution, and such knowledge is still lacking for many invasive species. Genus Ludwigia is characterized by a complex taxonomy, with an underexplored evolutionary history. Species from section Jussieae form a polyploid complex with diploids, tetraploids, hexaploids, and decaploids that are notorious invaders in freshwater and riparian ecosystems (Thouvenot et al.2013). Molecular phylogeny of the genus based on nuclear and chloroplast sequences (Liu et al. 2027) suggested some relationships between diploid and polyploid species, without fully resolving the question of the parentage of the polyploids. In their study, Barloy et al. (2023) have used a combination of molecular cytogenetics (Genomic In situ Hybridization), morphology and experimental crosses to elucidate the genomic compositions of the polyploid species, and show that the examined polyploids are of hybrid origin (allopolyploids). The tetraploid L. stolonifera derives from the diploids L. peploides subsp. montevidensis (AA genome) and L. helminthorhiza (BB genome). The tetraploid L. ascendens also share the BB genome combined with an undetermined different genome. The hexaploid L. grandiflora subsp. grandiflora has inherited the diploid AA genome combined with additional unidentified genomes. The decaploid L. grandiflora subsp. hexapetala has inherited the tetraploid L. stolonifera and the hexaploid L. grandiflora subsp. hexapetala genomes. As the authors point out, further work is needed, including additional related diploid (e.g. other subspecies of L. peploides) or tetraploid (L. hookeri and L. peduncularis) taxa that remain to be investigated, to address the nature of the undetermined parental genomes mentioned above. The presented work (Barloy et al. 2023) provides significant knowledge of this poorly investigated group with regard to genomic information and polyploid origin, and opens perspectives for future studies. The authors also detect additional diagnostic morphological traits of interest for in-situ discrimination of the taxa when monitoring invasive populations. References Barloy D., Portillo-Lemus L., Krueger-Hadfield S.A., Huteau V., Coriton O. (2024). Genomic relationships among diploid and polyploid species of the genus Ludwigia L. section Jussiaea using a combination of molecular cytogenetic, morphological, and crossing investigations. BioRxiv, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology https://doi.org/10.1101/2023.01.02.522458 te Beest M., Le Roux J.J., Richardson D.M., Brysting A.K., Suda J., Kubešová M., Pyšek P. (2012). The more the better? The role of polyploidy in facilitating plant invasions. Annals of Botany, Volume 109, Issue 1 Pages 19–45, https://doi.org/10.1093/aob/mcr277 Ramsey J. and Ramsey T. S. (2014). Ecological studies of polyploidy in the 100 years following its discovery Phil. Trans. R. Soc. B369 1–20 https://doi.org/10.1098/rstb.2013.0352 Rice, A., Šmarda, P., Novosolov, M. et al. (2019). The global biogeography of polyploid plants. Nat Ecol Evol 3, 265–273. https://doi.org/10.1038/s41559-018-0787-9 Thouvenot L, Haury J, Thiebaut G. (2013). A success story: Water primroses, aquatic plant pests. Aquat. Conserv. Mar. Freshw. Ecosyst. 23:790–803 https://doi.org/10.1002/aqc.2387 | Genomic relationships among diploid and polyploid species of the genus *Ludwigia* L. section *Jussiaea* using a combination of molecular cytogenetic, morphological, and crossing investigations | D. Barloy, L. Portillo - Lemus, S. A. Krueger-Hadfield, V. Huteau, O. Coriton | <p>ABSTRACTThe genus Ludwigia L. sectionJussiaeais composed of a polyploid species complex with 2x, 4x, 6x and 10x ploidy levels, suggesting possible hybrid origins. The aim of the present study is to understand the genomic relationships among dip... | | Hybridization / Introgression, Phylogenetics / Phylogenomics | Malika AINOUCHE | 2023-01-11 13:47:18 | ||
31 Oct 2022
Genotypic sex shapes maternal care in the African Pygmy mouse, Mus minutoidesEffect of sex chromosomes on mammalian behaviour: a case study in pygmy miceRecommended by Gabriel Marais and Trine Bilde based on reviews by Marion Anne-Lise Picard, Caroline Hu and 1 anonymous reviewer based on reviews by Marion Anne-Lise Picard, Caroline Hu and 1 anonymous reviewer
In mammals, it is well documented that sexual dimorphism and in particular sex differences in behaviour are fine-tuned by gonadal hormonal profiles. For example, in lemurs, where female social dominance is common, the level of testosterone in females is unusually high compared to that of other primate females (Petty and Drea 2015). Recent studies however suggest that gonadal hormones might not be the only biological factor involved in establishing sexual dimorphism, sex chromosomes might also play a role. The four core genotype (FCG) model and other similar systems allowing to decouple phenotypic and genotypic sex in mice have provided very convincing evidence of such a role (Gatewood et al. 2006; Arnold and Chen 2009; Arnold 2020a, 2020b). This however is a new field of research and the role of sex chromosomes in establishing sexually dimorphic behaviours has not been studied very much yet. Moreover, the FCG model has some limits. Sry, the male determinant gene on the mammalian Y chromosome might be involved in some sex differences in neuroanatomy, but Sry is always associated with maleness in the FCG model, and this potential role of Sry cannot be studied using this system. Heitzmann et al. have used a natural system to approach these questions. They worked on the African Pygmy mouse, Mus minutoides, in which a modified X chromosome called X* can feminize X*Y individuals, which offers a great opportunity for elegant experiments on the effects of sex chromosomes versus hormones on behaviour. They focused on maternal care and compared pup retrieval, nest quality, and mother-pup interactions in XX, X*X and X*Y females. They found that X*Y females are significantly better at retrieving pups than other females. They are also much more aggressive towards the fathers than other females, preventing paternal care. They build nests of poorer quality but have similar interactions with pups compared to other females. Importantly, no significant differences were found between XX and X*X females for these traits, which points to an effect of the Y chromosome in explaining the differences between X*Y and other females (XX, X*X). Also, another work from the same group showed similar gonadal hormone levels in all the females (Veyrunes et al. 2022). Heitzmann et al. made a number of predictions based on what is known about the neuroanatomy of rodents which might explain such behaviours. Using cytology, they looked for differences in neuron numbers in the hypothalamus involved in the oxytocin, vasopressin and dopaminergic pathways in XX, X*X and X*Y females, but could not find any significant effects. However, this part of their work relied on very small sample sizes and they used virgin females instead of mothers for ethical reasons, which greatly limited the analysis. Interestingly, X*Y females have a higher reproductive performance than XX and X*X ones, which compensate for the cost of producing unviable YY embryos and certainly contribute to maintaining a high frequency of X* in many African pygmy mice populations (Saunders et al. 2014, 2022). X*Y females are probably solitary mothers contrary to other females, and Heitzmann et al. have uncovered a divergent female strategy in this species. Their work points out the role of sex chromosomes in establishing sex differences in behaviours. References Arnold AP (2020a) Sexual differentiation of brain and other tissues: Five questions for the next 50 years. Hormones and Behavior, 120, 104691. https://doi.org/10.1016/j.yhbeh.2020.104691 Arnold AP (2020b) Four Core Genotypes and XY* mouse models: Update on impact on SABV research. Neuroscience & Biobehavioral Reviews, 119, 1–8. https://doi.org/10.1016/j.neubiorev.2020.09.021 Arnold AP, Chen X (2009) What does the “four core genotypes” mouse model tell us about sex differences in the brain and other tissues? Frontiers in Neuroendocrinology, 30, 1–9. https://doi.org/10.1016/j.yfrne.2008.11.001 Gatewood JD, Wills A, Shetty S, Xu J, Arnold AP, Burgoyne PS, Rissman EF (2006) Sex Chromosome Complement and Gonadal Sex Influence Aggressive and Parental Behaviors in Mice. Journal of Neuroscience, 26, 2335–2342. https://doi.org/10.1523/JNEUROSCI.3743-05.2006 Heitzmann LD, Challe M, Perez J, Castell L, Galibert E, Martin A, Valjent E, Veyrunes F (2022) Genotypic sex shapes maternal care in the African Pygmy mouse, Mus minutoides. bioRxiv, 2022.04.05.487174, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.04.05.487174 Petty JMA, Drea CM (2015) Female rule in lemurs is ancestral and hormonally mediated. Scientific Reports, 5, 9631. https://doi.org/10.1038/srep09631 Saunders PA, Perez J, Rahmoun M, Ronce O, Crochet P-A, Veyrunes F (2014) Xy Females Do Better Than the Xx in the African Pygmy Mouse, Mus Minutoides. Evolution, 68, 2119–2127. https://doi.org/10.1111/evo.12387 Saunders PA, Perez J, Ronce O, Veyrunes F (2022) Multiple sex chromosome drivers in a mammal with three sex chromosomes. Current Biology, 32, 2001-2010.e3. https://doi.org/10.1016/j.cub.2022.03.029 Veyrunes F, Perez J, Heitzmann L, Saunders PA, Givalois L (2022) Separating the effects of sex hormones and sex chromosomes on behavior in the African pygmy mouse Mus minutoides, a species with XY female sex reversal. bioRxiv, 2022.07.11.499546. https://doi.org/10.1101/2022.07.11.499546 | Genotypic sex shapes maternal care in the African Pygmy mouse, Mus minutoides | Louise D Heitzmann, Marie Challe, Julie Perez, Laia Castell, Evelyne Galibert, Agnes Martin, Emmanuel Valjent, Frederic Veyrunes | <p>Sexually dimorphic behaviours, such as parental care, have long been thought to be driven mostly, if not exclusively, by gonadal hormones. In the past two decades, a few studies have challenged this view, highlighting the direct influence of th... | | Behavior & Social Evolution, Evolutionary Ecology, Reproduction and Sex | Gabriel Marais | 2022-04-08 20:09:58 | ||
19 Dec 2016

POSTPRINT
Geographic body size variation in the periodical cicadas Magicicada: implications for life cycle divergence and local adaptationMegacicadas show a temperature-mediated converse Bergmann cline in body size (larger in the warmer south) but no body size difference between 13- and 17-year species pairsRecommended by Wolf Blanckenhorn and Thomas FlattPeriodical cicadas are a very prominent insect group in North America that are known for their large size, good looks, and loud sounds. However, they are probably known best to evolutionary ecologists because of their long juvenile periods of 13 or 17 years (prime numbers!), which they spend in the ground. Multiple related species living in the same area are often coordinated in emerging as adults during the same year, thereby presumably swamping any predators specialized on eating them. Reference [1] Koyama T, Ito H, Kakishima S, Yoshimura J, Cooley JR, Simon C, Sota T. 2015. Geographic body size variation in the periodical cicadas Magicicada: implications for life cycle divergence and local adaptation. Journal of Evolutionary Biology 28:1270-1277. doi: 10.1111/jeb.12653 | Geographic body size variation in the periodical cicadas Magicicada: implications for life cycle divergence and local adaptation | Koyama T, Ito H, Kakishima S, Yoshimura J, Cooley JR, Simon C, Sota T | Seven species in three species groups (Decim, Cassini and Decula) of periodical cicadas (*Magicicada*) occupy a wide latitudinal range in the eastern United States. To clarify how adult body size, a key trait affecting fitness, varies geographical... | | Adaptation, Evolutionary Ecology, Life History, Macroevolution, Phylogeography & Biogeography, Speciation | Wolf Blanckenhorn | 2016-12-19 10:39:22 | ||
22 Oct 2019

Geographic variation in adult and embryonic desiccation tolerance in a terrestrial-breeding frogTough as old boots: amphibians from drier habitats are more resistant to desiccation, but less flexible at exploiting wet conditionsRecommended by Ben Phillips based on reviews by Juan Diego Gaitan-Espitia, Jennifer Nicole Lohr and 1 anonymous reviewerSpecies everywhere are facing rapid climatic change, and we are increasingly asking whether populations will adapt, shift, or perish [1]. There is a growing realisation that, despite limited within-population genetic variation, many species exhibit substantial geographic variation in climate-relevant traits. This geographic variation might play an important role in facilitating adaptation to climate change [2,3]. References [1] Hoffmann, A. A., and Sgrò, C. M. (2011). Climate change and evolutionary adaptation. Nature, 470(7335), 479–485. doi: 10.1038/nature09670 | Geographic variation in adult and embryonic desiccation tolerance in a terrestrial-breeding frog | Rudin-Bitterli, T, Evans, J. P. and Mitchell, N. J. | <p>Intra-specific variation in the ability of individuals to tolerate environmental perturbations is often neglected when considering the impacts of climate change. Yet this information is potentially crucial for mitigating any deleterious effects... | | Adaptation, Evolutionary Applications, Evolutionary Ecology | Ben Phillips | 2018-05-07 03:35:08 | ||
02 Feb 2023
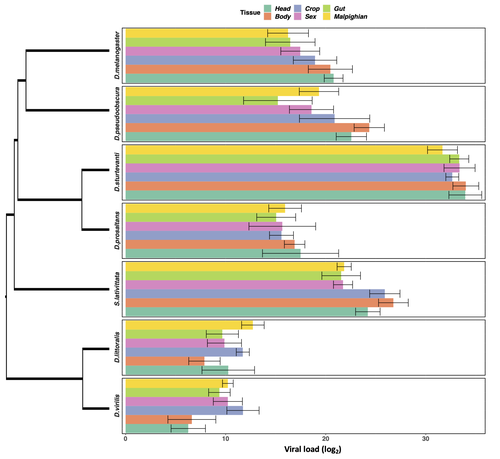
Heterogeneities in infection outcomes across species: sex and tissue differences in virus susceptibilitySusceptibility to infection is not explained by sex or differences in tissue tropism across different species of DrosophilaRecommended by Alison Duncan based on reviews by Greg Hurst and 1 anonymous reviewerUnderstanding factors explaining both intra and interspecific variation in susceptibility to infection by parasites remains a key question in evolutionary biology. Within a species variation in susceptibility is often explained by differences in behaviour affecting exposure to infection and/or resistance affecting the degree by which parasite growth is controlled (Roy & Kirchner, 2000, Behringer et al., 2000). This can vary between the sexes (Kelly et al., 2018) and may be explained by the ability of a parasite to attack different organs or tissues (Brierley et al., 2019). However, what goes on within one species is not always relevant to another, making it unclear when patterns can be scaled up and generalised across species. This is also important to understand when parasites may jump hosts, or identify species that may be susceptible to a host jump (Longdon et al., 2015). Phylogenetic distance between hosts is often an important factor explaining susceptibility to a particular parasite in plant and animal hosts (Gilbert & Webb, 2007, Faria et al., 2013). In two separate experiments, Roberts and Longdon (Roberts & Longdon, 2022) investigated how sex and tissue tropism affected variation in the load of Drosophila C Virus (DCV) across multiple Drosophila species. DCV load has been shown to correlate positively with mortality (Longdon et al., 2015). Overall, they found that load did not vary between the sexes; within a species males and females had similar DCV loads for 31 different species. There was some variation in levels of DCV growth in different tissue types, but these too were consistent across males for 7 species of Drosophila. Instead, in both experiments, host phylogeny or interspecific variation, explained differences in DCV load with some species being more infected than others. This study is neat in that it incorporates and explores simultaneously both intra and interspecific variation in infection-related life-history traits which is not often done (but see (Longdon et al., 2015, Imrie et al., 2021, Longdon et al., 2011, Johnson et al., 2012). Indeed, most studies to date explore either inter-specific differences in susceptibility to a parasite (it can or can’t infect a given species) (Davies & Pedersen, 2008, Pfenning-Butterworth et al., 2021) or intra-specific variability in infection-related traits (infectivity, resistance etc.) due to factors such as sex, genotype and environment (Vale et al., 2008, Lambrechts et al., 2006). This work thus advances on previous studies, while at the same time showing that sex differences in parasite load are not necessarily pervasive. References Behringer DC, Butler MJ, Shields JD (2006) Avoidance of disease by social lobsters. Nature, 441, 421–421. https://doi.org/10.1038/441421a Brierley L, Pedersen AB, Woolhouse MEJ (2019) Tissue tropism and transmission ecology predict virulence of human RNA viruses. PLOS Biology, 17, e3000206. https://doi.org/10.1371/journal.pbio.3000206 Davies TJ, Pedersen AB (2008) Phylogeny and geography predict pathogen community similarity in wild primates and humans. Proceedings of the Royal Society B: Biological Sciences, 275, 1695–1701. https://doi.org/10.1098/rspb.2008.0284 Faria NR, Suchard MA, Rambaut A, Streicker DG, Lemey P (2013) Simultaneously reconstructing viral cross-species transmission history and identifying the underlying constraints. Philosophical Transactions of the Royal Society B: Biological Sciences, 368, 20120196. https://doi.org/10.1098/rstb.2012.0196 Gilbert GS, Webb CO (2007) Phylogenetic signal in plant pathogen–host range. Proceedings of the National Academy of Sciences, 104, 4979–4983. https://doi.org/10.1073/pnas.0607968104 Imrie RM, Roberts KE, Longdon B (2021) Between virus correlations in the outcome of infection across host species: Evidence of virus by host species interactions. Evolution Letters, 5, 472–483. https://doi.org/10.1002/evl3.247 Johnson PTJ, Rohr JR, Hoverman JT, Kellermanns E, Bowerman J, Lunde KB (2012) Living fast and dying of infection: host life history drives interspecific variation in infection and disease risk. Ecology Letters, 15, 235–242. https://doi.org/10.1111/j.1461-0248.2011.01730.x Kelly CD, Stoehr AM, Nunn C, Smyth KN, Prokop ZM (2018) Sexual dimorphism in immunity across animals: a meta-analysis. Ecology Letters, 21, 1885–1894. https://doi.org/10.1111/ele.13164 Lambrechts L, Chavatte J-M, Snounou G, Koella JC (2006) Environmental influence on the genetic basis of mosquito resistance to malaria parasites. Proceedings of the Royal Society B: Biological Sciences, 273, 1501–1506. https://doi.org/10.1098/rspb.2006.3483 Longdon B, Hadfield JD, Day JP, Smith SCL, McGonigle JE, Cogni R, Cao C, Jiggins FM (2015) The Causes and Consequences of Changes in Virulence following Pathogen Host Shifts. PLOS Pathogens, 11, e1004728. https://doi.org/10.1371/journal.ppat.1004728 Longdon B, Hadfield JD, Webster CL, Obbard DJ, Jiggins FM (2011) Host Phylogeny Determines Viral Persistence and Replication in Novel Hosts. PLOS Pathogens, 7, e1002260. https://doi.org/10.1371/journal.ppat.1002260 Pfenning-Butterworth AC, Davies TJ, Cressler CE (2021) Identifying co-phylogenetic hotspots for zoonotic disease. Philosophical Transactions of the Royal Society B: Biological Sciences, 376, 20200363. https://doi.org/10.1098/rstb.2020.0363 Roberts KE, Longdon B (2023) Heterogeneities in infection outcomes across species: examining sex and tissue differences in virus susceptibility. bioRxiv 2022.11.01.514663, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.11.01.514663 Roy BA, Kirchner JW (2000) Evolutionary Dynamics of Pathogen Resistance and Tolerance. Evolution, 54, 51–63. https://doi.org/10.1111/j.0014-3820.2000.tb00007.x Vale PF, Stjernman M, Little TJ (2008) Temperature-dependent costs of parasitism and maintenance of polymorphism under genotype-by-environment interactions. Journal of Evolutionary Biology, 21, 1418–1427. https://doi.org/10.1111/j.1420-9101.2008.01555.x | Heterogeneities in infection outcomes across species: sex and tissue differences in virus susceptibility | Katherine E Roberts, Ben Longdon | <p style="text-align: justify;">Species vary in their susceptibility to pathogens, and this can alter the ability of a pathogen to infect a novel host. However, many factors can generate heterogeneity in infection outcomes, obscuring our ability t... | | Evolutionary Ecology | Alison Duncan | Anonymous, Greg Hurst | 2022-11-03 11:17:42 | |
14 Dec 2016
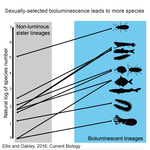
POSTPRINT
High Rates of Species Accumulation in Animals with Bioluminescent Courtship DisplaysBioluminescent sexually selected traits as an engine for biodiversity across animal speciesRecommended by Astrid Groot and Carole SmadjaIn evolutionary biology, sexual selection is hypothesized to increase speciation rates in animals, as theory predicts that sexual selection will contribute to phenotypic diversification and affect rates of species accumulation at macro-evolutionary time scales. However, testing this hypothesis and gathering convincing evidence have proven difficult. Although some studies have shown a strong correlation between proxies of sexual selection and species diversity (mostly in birds), this relationship relies on some assumptions on the link between these proxies and the strength of sexual selection and is not detected in some other taxa, making taxonomically widespread conclusions impossible. In a recent study published in Current Biology [1], Ellis and Oakley provide strong evidence that bioluminescent sexual displays have driven high species richness in taxonomically diverse animal lineages, providing a crucial link between sexual selection and speciation. Ellis and Oakley [1] explored the scientific literature for well-resolved evolutionary trees with branches containing bioluminescent lineages and identified lineages that use light for courtship or camouflage in a wide range of marine and terrestrial taxa including insects, crustaceans, cephalopods, segmented worms, and fishes. The researchers counted the number of species in each bioluminescent clade and found that all groups with light-courtship displays had more species and faster rates of species accumulation than their non-luminous most closely related sister lineages or ancestors. In contrast, those groups that used bioluminescence for predator avoidance had a lower than expected rate of species richness on average. Nicely encompassing a diversity of taxa and neatly controlling for the rate of species accumulation of the encompassing clade, the results of Ellis and Oakley are clear-cut and provide the most comprehensive evidence to date for the hypothesis that sexual displays can act as drivers of speciation. One question this study incites is what is happening in terms of sexual selection in species displaying defensive bioluminescence or no bioluminescence at all: do those lineages use no mating signals at all or other mating signals that are less apparent, and will those experience lower levels of sexual selection than bioluminescent mating signals, i.e. consistent with Ellis and Oakley results? It would also be interesting to investigate the diversification rates in animal species using other modalities, such as chemical, acoustic or any other type of signals used by males, females or both sexes, to determine what types of sexual signals may be more generally drivers of speciation. References [1] Ellis EA, Oakley TH. 2016. High Rates of Species Accumulation in Animals with Bioluminescent Courtship Displays. Current Biology 26:1916–1921. doi: 10.1016/j.cub.2016.05.043 [2] Davis MP, Holcroft NI, Wiley EO, Sparks JS, Smith WL. 2014. Species-specific bioluminescence facilitates speciation in the deep sea. Marine Biology 161:11391148. doi: 10.1007/s00227-014-2406-x [3] Davis MP, Sparks JS, Smith WL. 2016. Repeated and Widespread Evolution of Bioluminescence in Marine Fishes. PLoS One 11:e0155154. doi: 10.1371/journal.pone.0155154 [4] Claes JM, Nilsson D-E, Mallefet J, Straube N. 2015. The presence of lateral photophores correlates with increased speciation in deep-sea bioluminescent sharks. Royal Society Open Science 2:150219. doi: 10.1098/rsos.150219 | High Rates of Species Accumulation in Animals with Bioluminescent Courtship Displays | Ellis EA, Oakley TH | One of the great mysteries of evolutionary biology is why closely related lineages accumulate species at different rates. Theory predicts that populations undergoing strong sexual selection will more quickly differentiate because of increased pote... | | Adaptation, Evolutionary Ecology, Sexual Selection, Speciation | Astrid Groot | 2016-12-14 19:01:59 | ||
19 Jul 2021

Host phenology can drive the evolution of intermediate virulence strategies in some obligate-killer parasitesModelling parasitoid virulence evolution with seasonalityRecommended by Samuel Alizon based on reviews by Alex Best and 2 anonymous reviewers
The harm most parasites cause to their host, i.e. the virulence, is a mystery because host death often means the end of the infectious period. For obligate killer parasites, or “parasitoids”, that need to kill their host to transmit to other hosts the question is reversed. Indeed, more rapid host death means shorter generation intervals between two infections and mathematical models show that, in the simplest settings, natural selection should always favour more virulent strains (Levin and Lenski, 1983). Adding biological details to the model modifies this conclusion and, for instance, if the relationship between the infection duration and the number of parasites transmission stages produced in a host is non-linear, strains with intermediate levels of virulence can be favoured (Ebert and Weisser 1997). Other effects, such as spatial structure, could yield similar effects (Lion and van Baalen, 2007). In their study, MacDonald et al. (2021) explore another type of constraint, which is seasonality. Earlier studies, such as that by Donnelly et al. (2013) showed that this constraint can affect virulence evolution but they had focused on directly transmitted parasites. Using a mathematical model capturing the dynamics of a parasitoid, MacDonald et al. (2021) show if two main assumptions are met, namely that at the end of the season only transmission stages (or “propagules”) survive and that there is a constant decay of these propagules with time, then strains with intermediate levels of virulence are favoured. Practically, the authors use delay differential equations and an adaptive dynamics approach to identify evolutionary stable strategies. As expected, the longer the short the season length, the higher the virulence (because propagule decay matters less). The authors also identify a non-linear relationship between the variation in host development time and virulence. Generally, the larger the variation, the higher the virulence because the parasitoid has to kill its host before the end of the season. However, if the variation is too wide, some hosts become physically impossible to use for the parasite, whence a decrease in virulence. Finally, MacDonald et ali. (2021) show that the consequence of adding trade-offs between infection duration and the number of propagules produced is in line with earlier studies (Ebert and Weisser 1997). These mathematical modelling results provide testable predictions for using well-described systems in evolutionary ecology such as daphnia parasitoids, baculoviruses, or lytic phages. Reference Donnelly R, Best A, White A, Boots M (2013) Seasonality selects for more acutely virulent parasites when virulence is density dependent. Proc R Soc B, 280, 20122464. https://doi.org/10.1098/rspb.2012.2464 Ebert D, Weisser WW (1997) Optimal killing for obligate killers: the evolution of life histories and virulence of semelparous parasites. Proc R Soc B, 264, 985–991. https://doi.org/10.1098/rspb.1997.0136 Levin BR, Lenski RE (1983) Coevolution in bacteria and their viruses and plasmids. In: Futuyma DJ, Slatkin M eds. Coevolution. Sunderland, MA, USA: Sinauer Associates, Inc., 99–127. Lion S, van Baalen M (2008) Self-structuring in spatial evolutionary ecology. Ecol. Lett., 11, 277–295. https://doi.org/10.1111/j.1461-0248.2007.01132.x MacDonald H, Akçay E, Brisson D (2021) Host phenology can drive the evolution of intermediate virulence strategies in some obligate-killer parasites. bioRxiv, 2021.03.13.435259, ver. 8 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.03.13.435259 | Host phenology can drive the evolution of intermediate virulence strategies in some obligate-killer parasites | Hannelore MacDonald, Erol Akçay, Dustin Brisson | <p style="text-align: justify;">The traditional mechanistic trade-offs resulting in a negative correlation between transmission and virulence are the foundation of nearly all current theory on the evolution of parasite virulence. Several ecologica... | | Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Epidemiology, Evolutionary Theory | Samuel Alizon | 2021-03-14 13:47:33 | ||
02 May 2023
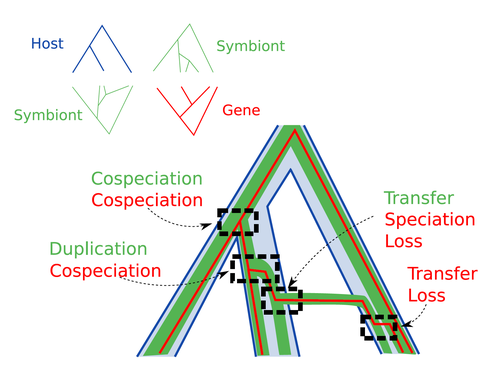
Host-symbiont-gene phylogenetic reconciliationReconciling molecular evolution and evolutionary ecology studies: a phylogenetic reconciliation method for gene-symbiont-host systemsRecommended by Emmanuelle Jousselin based on reviews by Vincent Berry and Catherine MatiasInteractions between species are a driving force in evolution. Many organisms host symbiotic partners that live all or part of their life in or on their host. Whether they are mutualistic or parasitic, these symbiotic associations impose strong selective pressures on both partners and affect their evolutionary trajectories. In fine, they can have a significant impact on the diversification patterns of both host and symbiont lineages, with symbiotic lineages sometimes speciating simultaneously with their hosts and/or switching from one host species to another. Long-term associations between species can also result in gene transfers between the involved organisms. Those lateral gene transfers are a source of ecological innovation but can obscure the phylogenetic signals and render the process of phylogenetic reconstructions complex (Lerat et al. 2003). Methods known as reconciliations explore similarities and differences between phylogenetic trees. They have been widely used to both compare the diversification patterns of hosts and symbionts and identify lateral gene transfers between species. Though the reconciliation approaches used in host/ symbiont and species/ gene phylogenetic studies are identical, they are always applied separately to solve either molecular evolution questions or investigate the evolution of ecological interactions. However, the two questions are often intimately linked and the current interest in multi-level systems (e.g. the holobiont concept) calls for a unique model that will take into account three-level nested organization (gene/symbiont/ host) where both symbiont and genes can transfer among hosts. Here Menet and collaborators (2023) provide such a model to produce three-level reconciliations. In order to do so, they extend the two-level reconciliation model implemented in “ALE” software (Szöllősi et al. 2013), one of the most used and proven reconciliation methods. Briefly, given a symbiont gene tree, a symbiont tree and a host tree, as in previous reconciliation models, the symbiont tree is mapped onto the host tree by mixing three types of events: Duplication, Transfer or Loss (DTL), with a possibility of the symbiont evolving temporarily outside the host phylogeny (in a “ghost” host lineage). The gene tree evolves similarly inside the symbiont tree, but horizontal transfers are constrained to symbionts co-occurring within the same host. Joint reconciliation scenarios are reconstructed and DTL event rates and likelihoods are estimated according to the model. As a nice addition, the authors propose a method to infer the symbiont phylogeny through amalgamation from gene trees and a host tree. The authors then explore the diverse possibilities offered by this method by testing it on both simulated datasets and biological datasets in order to check whether considering three nested levels is worthwhile. They convincingly show that three-level reconciliation has a better capacity to retrieve the symbiont donors and receivers of horizontal gene transfers, probably because transfers are constrained by additional elements relevant to the biological systems. Using, aphids, their obligate endosymbionts, and the symbiont genes involved in their nutritional functions, they identify horizontal gene transfers between aphid symbionts that are missed by two-level reconciliations but detected by expertise (Manzano-Marín et al. 2020). The other dataset presented here is on the human pathogen Helicobacter pylori, which history is supposed to reflect human migration. They use more than 1000 H. pylori gene families, and four populations, and use likelihood computations to compare different hypotheses on the diversification of the host. In summary, this study is a proof-of-concept of a 3-level reconciliation, where the authors manage to convey the applicability of their framework to many biological systems. Reported complexities, confirmed by reported running times, show that the method is computationally efficient. Without a doubt, the tool presented here will be very useful to evolutionary biologists who want to investigate multi-scale cophylogenies and it will move forward the study of associations between host and symbionts when symbiont genomic data are available. REFERENCES Lerat, E., Daubin, V., & Moran, N. A. (2003). From gene trees to organismal phylogeny in prokaryotes: the case of the γ-Proteobacteria. PLoS biology, 1(1), e19. Menet H, Trung AN, Daubin V, Tannier E (2023) Host-symbiont-gene phylogenetic reconciliation. bioRxiv, 2022.07.01.498457, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.07.01.498457 Szöllősi, G. J., Rosikiewicz, W., Boussau, B., Tannier, E., & Daubin, V. (2013). Efficient exploration of the space of reconciled gene trees. Systematic biology, 62(6), 901-912. | Host-symbiont-gene phylogenetic reconciliation | Hugo Menet, Alexia Nguyen Trung, Vincent Daubin, Eric Tannier | <p style="text-align: justify;"><strong>Motivation:</strong> Biological systems are made of entities organized at different scales e.g. macro-organisms, symbionts, genes...) which evolve in interaction.<br>These interactions range from indepe... | | Bioinformatics & Computational Biology, Phylogenetics / Phylogenomics | Emmanuelle Jousselin | 2022-08-21 18:34:27 |




