
Latest recommendations

| Id | Title * | Authors * | Abstract * | Picture * ▲ | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
20 Nov 2017
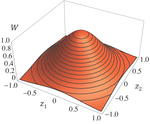
Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selectionUnderstanding genetic variance, load, and inbreeding depression with selfingRecommended by Aneil F. Agrawal based on reviews by Frédéric Guillaume and 1 anonymous reviewerA classic problem in evolutionary biology is to understand the genetic variance in fitness. The simplest hypothesis is that variation exists, even in well-adapted populations, as a result of the balance between mutational input and selective elimination. This variation causes a reduction in mean fitness, known as the mutation load. Though mutation load is difficult to quantify empirically, indirect evidence of segregating genetic variation in fitness is often readily obtained by comparing the fitness of inbred and outbred offspring, i.e., by measuring inbreeding depression. Mutation-selection balance models have been studied as a means of understanding the genetic variance in fitness, mutation load, and inbreeding depression. Since their inception, such models have increased in sophistication, allowing us to ask these questions under more realistic and varied scenarios. The new theoretical work by Abu Awad and Roze [1] is a substantial step forward in understanding how arbitrary levels of self-fertilization affect variation, load and inbreeding depression under mutation-selection balance. References [1] Abu Awad D and Roze D. 2017. Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selection. bioRxiv, 180000, ver. 4 of 17th November 2017. doi: 10.1101/180000 [2] Lande R. 1977. The influence of the mating system on the maintenance of genetic variability in polygenic characters. Genetics 86: 485–498. [3] Charlesworth D and Charlesworth B. 1987. Inbreeding depression and its evolutionary consequences. Annual Review of Ecology and Systematics. 18: 237–268. doi: 10.1111/10.1146/annurev.es.18.110187.001321 [4] Lande R and Porcher E. 2015. Maintenance of quantitative genetic variance under partial self-fertilization, with implications for the evolution of selfing. Genetics 200: 891–906. doi: 10.1534/genetics.115.176693 [5] Roze D. 2015. Effects of interference between selected loci on the mutation load, inbreeding depression, and heterosis. Genetics 201: 745–757. doi: 10.1534/genetics.115.178533 [6] Martin G and Lenormand T. 2006. A general multivariate extension of Fisher's geometrical model and the distribution of mutation fitness effects across species. Evolution 60: 893–907. doi: 10.1111/j.0014-3820.2006.tb01169.x [7] Martin G, Elena SF and Lenormand T. 2007. Distributions of epistasis in microbes fit predictions from a fitness landscape model. Nature Genetics 39: 555–560. doi: 10.1038/ng1998 | Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selection | Diala Abu Awad and Denis Roze | The mating system of a species is expected to have important effects on its genetic diversity. In this paper, we explore the effects of partial selfing on the equilibrium genetic variance Vg, mutation load L and inbreeding depression δ under stabi... | | Evolutionary Theory, Population Genetics / Genomics, Quantitative Genetics, Reproduction and Sex | Aneil F. Agrawal | 2017-08-26 09:29:20 | ||
20 Dec 2016

POSTPRINT
Experimental Evolution of Gene Expression and Plasticity in Alternative Selective RegimesGenetic adaptation counters phenotypic plasticity in experimental evolutionRecommended by Luis-Miguel Chevin and Stephanie BedhommeHow do phenotypic plasticity and adaptive evolution interact in a novel or changing environment? Does evolution by natural selection generally reinforce initially plastic phenotypic responses, or does it instead oppose them? And to what extent does evolution of a trait involve evolution of its plasticity? These questions have lied at the heart of research on phenotypic evolution in heterogeneous environments ever since it was realized that the environment is likely to affect the expression of many (perhaps most) characters of an individual. Importantly, this broad definition of phenotypic plasticity as change in the average phenotype of a given genotype in response to its environment of development (or expression) does not involve any statement about the adaptiveness of the plastic changes. Theory on the evolution of plasticity has devoted much effort to understanding how reaction norm should evolve under different regimes of environmental change in space and time, and depending on genetic constraints on reaction norm shapes. However on an empirically ground, the questions above have mostly been addressed for individual traits, often chosen a priori for their likeliness to exhibit adaptive plasticity, and we still lack more systematic answers. These can be provided by so-called ‘phenomic’ approaches, where a large number of traits are tracked without prior information on their biological or ecological function. A problem is that the number of phenotypic characters that can be measured in an organism is virtually infinite (and to some extent arbitrary), and that scaling issues makes it difficult to compare different sets of traits. Gene-expression levels offer a partial solution to this dilemma, as they can be considered as a very large number of traits (one per typed gene) that can be measured easily and uniformly (fold change in the number of reads in RNAseq). As for any traits, expression levels of different genes may be genetically correlated, to an extent that depends on their regulation mechanism: cis-regulatory sequences that only affect expression of neighboring genes are likely to cause independent gene expression, while more systematic modifiers of expression (e.g. trans-regulators such as transcription factors) may cause correlated genetic responses of the expression of many genes. Huang and Agrawal [1] have studied plasticity and evolution of gene expression level in young larvae of populations of Drosophila melanogaster that have evolved for about 130 generations under either a constant environment (salt or cadmium), or an environment that is heterogeneous in time or space (combining salt and cadmium). They report a wealth of results, of which we summarize the most striking here. First, among genes that (i) were initially highly plastic and (ii) evolved significant divergence in expression levels between constant environment treatments, the evolved divergence is predominantly in the opposite direction to the initial plastic response. This suggests that either plasticity was initially maladaptive, or the selective pressure changed during the evolutionary process (see below). This somewhat unexpected result strikingly mirrors that from a study published last year in Nature [2], where the same pattern was found for responses of guppies to the presence of predators. However, Huang and Agrawal [1] went beyond this study by deciphering the underlying mechanisms in several interesting ways. First, they showed that change in gene expression often occurred at genes close to SNPs with differentiated frequencies across treatments (but not at genes with differentiated SNPs in their coding sequences), suggesting that cis-regulatory sequences are involved. This is also suggested by the fact that changes in gene expression are mostly caused by the increased expression of only one allele at polymorphic loci, and is a first step towards investigating the genetic underpinnings of (co)variation in gene expression levels. Another interesting set of findings concerns evolution of plasticity in treatments with variable environments. To compare the gene-expression plasticity that evolved in these treatments to an expectation, the authors considered that the expression levels in populations maintained for a long time under constant salt or cadmium had reached an optimum. The differences between these expression levels were thus assumed to predict the level of plasticity that should evolve in a heterogeneous environment (with both cadmium and salt) under perfect environmental predictability. The authors showed that plasticity did evolve more in the expected direction in heterogeneous than in constant environments, resulting in better adapted final expression levels across environments. Taken collectively, these results provide an unprecedented set of patterns that are greatly informative on how plasticity and evolution interact in constant versus changing environments. But of course, interpretations in terms of adaptive versus maladaptive plasticity are more challenging, as the authors themselves admit. Even though environmentally determined gene expression is the basic mechanism underlying the phenotypic plasticity of most traits, it is extremely difficult to relate to more integrated phenotypes for which we can understand the selection pressures, especially in multicellular organisms. The authors have recently investigated evolutionary change of quantitative traits in these selected lines, so it might be possible to establish links between reaction norms for macroscopic traits to those for gene expression levels. Such an approach would also involve tracking gene expression throughout life, rather than only in young larvae as done here, thus putting phenotypic complexity back in the picture also for expression levels. Another difficulty is that a plastic response that was originally adaptive may be replaced by an opposite evolutionary response in the long run, without having to invoke initially maladaptive plasticity. For instance, the authors mention the possibility that a generic stress response is initially triggered by cadmium, but is eventually unnecessary and costly after evolution of genetic mechanisms for cadmium detoxification (a case of so-called genetic accommodation). In any case, this study by Huang and Agrawal [1], together with the one by Ghalambor et al. last year [2], reports novel and unexpected results, which are likely to stimulate researchers interested in plasticity and evolution in heterogeneous environments for the years to come. References [1] Huang Y, Agrawal AF. 2016. Experimental Evolution of Gene Expression and Plasticity in Alternative Selective Regimes. PLoS Genetics 12:e1006336. doi: 10.1371/journal.pgen.1006336 [2] Ghalambor CK, Hoke KL, Ruell EW, Fischer EK, Reznick DN, Hughes KA. 2015. Non-adaptive plasticity potentiates rapid adaptive evolution of gene expression in nature. Nature 525: 372-375. doi: 10.1038/nature15256 | Experimental Evolution of Gene Expression and Plasticity in Alternative Selective Regimes | Huang Y, Agrawal AF | Little is known of how gene expression and its plasticity evolves as populations adapt to different environmental regimes. Expression is expected to evolve adaptively in all populations but only those populations experiencing environmental heterog... | | Adaptation, Experimental Evolution, Expression Studies, Phenotypic Plasticity | Luis-Miguel Chevin | 2016-12-20 09:04:15 | ||
22 Sep 2020
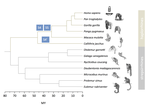
Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primatesStudying genetic antagonisms as drivers of genome evolutionRecommended by Mathieu Joron based on reviews by Qi Zhou and 3 anonymous reviewersSex chromosomes are special in the genome because they are often highly differentiated over much of their lengths and marked by degenerative evolution of their gene content. Understanding why sex chromosomes differentiate requires deciphering the forces driving their recombination patterns. Suppression of recombination may be subject to selection, notably because of functional effects of locking together variation at different traits, as well as longer-term consequences of the inefficient purge of deleterious mutations, both of which may contribute to patterns of differentiation [1]. As an example, male and female functions may reveal intrinsic antagonisms over the optimal genotypes at certain genes or certain combinations of interacting genes. As a result, selection may favour the recruitment of rearrangements blocking recombination and maintaining the association of sex-antagonistic allele combinations with the sex-determining locus. References [1] Charlesworth D (2017) Evolution of recombination rates between sex chromosomes. Philosophical Transactions of the Royal Society B: Biological Sciences, 372, 20160456. https://doi.org/10.1098/rstb.2016.0456 | Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primates | Rylan Shearn, Alison E. Wright, Sylvain Mousset, Corinne Régis, Simon Penel, Jean-François Lemaitre, Guillaume Douay, Brigitte Crouau-Roy, Emilie Lecompte, Gabriel A.B. Marais | <p>Sex chromosomes are typically comprised of a non-recombining region and a recombining pseudoautosomal region. Accurately quantifying the relative size of these regions is critical for sex chromosome biology both from a functional (i.e. number o... | | Bioinformatics & Computational Biology, Genome Evolution, Molecular Evolution, Reproduction and Sex, Sexual Selection | Mathieu Joron | 2019-02-04 15:16:32 | ||
14 Feb 2024
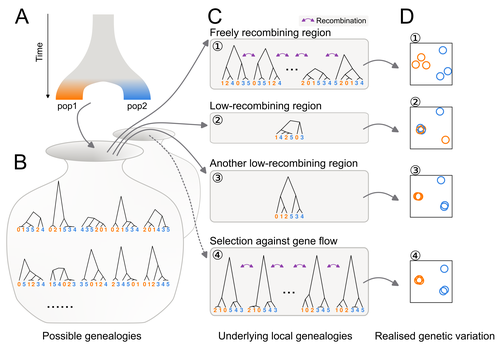
Distinct patterns of genetic variation at low-recombining genomic regions represent haplotype structureDiscerning the causes of local deviations in genetic variation: the effect of low-recombination regionsRecommended by Matteo Fumagalli based on reviews by Claire Merot and 1 anonymous reviewer based on reviews by Claire Merot and 1 anonymous reviewer
In this study, Ishigohoka and colleagues tackle an important, yet often overlooked, question on the causes of genetic variation. While genome-wide patterns represent population structure, local variation is often associated with selection. Authors propose that an alternative cause for variation in individual loci is reduced recombination rate. To test this hypothesis, authors perform local Principal Component Analysis (PCA) (Li & Ralph, 2019) to identify local deviations in population structure in the Eurasian blackcap (Sylvia atricapilla) (Ishigohoka et al. 2022). This approach is typically used to detect chromosomal rearrangements or any long region of linked loci (e.g., due to reduced recombination or selection) (Mérot et al. 2021). While other studies investigated the effect of low recombination on genetic variation (Booker et al. 2020), here authors provide a comprehensive analysis of the effect of recombination to local PCA patterns both in empirical and simulated data sets. Findings demonstrate that low recombination (and not selection) can be the sole explanatory variable for outlier windows. The study also describes patterns of genetic variation along the genome of Eurasian blackcaps, localising at least two polymorphic inversions (Ishigohoka et al. 2022). Further investigations on the effect of model parameters (e.g., window sizes and thresholds for defining low-recombining regions), as well as the use of powerful neutrality tests are in need to clearly assess whether outlier regions experience selection and reduced recombination, and to what extent. References Booker, T. R., Yeaman, S., & Whitlock, M. C. (2020). Variation in recombination rate affects detection of outliers in genome scans under neutrality. Molecular Ecology, 29 (22), 4274–4279. https://doi.org/10.1111/mec.15501 Ishigohoka, J., Bascón-Cardozo, K., Bours, A., Fuß, J., Rhie, A., Mountcastle, J., Haase, B., Chow, W., Collins, J., Howe, K., Uliano-Silva, M., Fedrigo, O., Jarvis, E. D., Pérez-Tris, J., Illera, J. C., Liedvogel, M. (2022) Distinct patterns of genetic variation at low-recombining genomic regions represent haplotype structure. bioRxiv 2021.12.22.473882, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.12.22.473882 Li, H., & Ralph, P. (2019). Local PCA Shows How the Effect of Population Structure Differs Along the Genome. Genetics, 211 (1), 289–304. https://doi.org/10.1534/genetics.118.301747 Mérot, C., Berdan, E. L., Cayuela, H., Djambazian, H., Ferchaud, A.-L., Laporte, M., Normandeau, E., Ragoussis, J., Wellenreuther, M., & Bernatchez, L. (2021). Locally Adaptive Inversions Modulate Genetic Variation at Different Geographic Scales in a Seaweed Fly. Molecular Biology and Evolution, 38 (9), 3953–3971. https://doi.org/10.1093/molbev/msab143 | Distinct patterns of genetic variation at low-recombining genomic regions represent haplotype structure | Jun Ishigohoka, Karen Bascón-Cardozo, Andrea Bours, Janina Fuß, Arang Rhie, Jacquelyn Mountcastle, Bettina Haase, William Chow, Joanna Collins, Kerstin Howe, Marcela Uliano-Silva, Olivier Fedrigo, Erich D. Jarvis, Javier Pérez-Tris, Juan Carlos Il... | <p>Genetic variation of the entire genome represents population structure, yet individual loci can show distinct patterns. Such deviations identified through genome scans have often been attributed to effects of selection instead of randomness. Th... | | Genome Evolution, Molecular Evolution, Population Genetics / Genomics | Matteo Fumagalli | 2023-10-13 11:58:47 | ||
05 Jun 2018
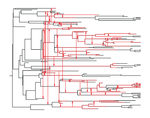
The dynamics of preferential host switching: host phylogeny as a key predictor of parasite prevalence and distributionShift or stick? Untangling the signatures of biased host switching, and host-parasite co-speciationRecommended by Lucy Weinert based on reviews by Damien de Vienne and Nathan MeddMany emerging diseases arise by parasites switching to new host species, while other parasites seem to remain with same host lineage for very long periods of time, even over timescales where an ancestral host species splits into two or more new species. The ability to understand these dynamics would form an important part of our understanding of infectious disease. Experiments are clearly important for understanding these processes, but so are comparative studies, investigating the variation that we find in nature. Such comparative data do show strong signs of non-randomness, and this suggests that the epidemiological and ecological processes might be predictable, at least in part. For example, when we map patterns of parasite presence/absence onto host phylogenies, we often find that certain host clades harbour many more parasites than expected, or that closely-related hosts harbour closely-related parasites. Nevertheless, it remains difficult to interpret these patterns to make inferences about ecological and epidemiological processes. This is partly because non-random associations can arise in multiple ways. For example, parasites might be inherited from the common ancestor of related hosts, or might switch to new hosts, but preferentially establish on novel hosts that are closely related to their existing host. Infection might also influence the shape of host phylogeny, either by increasing the rate of host extinction or, conversely, increasing the rate of speciation (as with manipulative symbionts that might induce reproductive isolation). These various processes have, by and large, been studied in isolation, but the model introduced by Engelstädter and Fortuna [1], makes an important first step towards studying them together. Without such combined analyses, we will not be able to tell if the processes have their own unique signatures, or whether the same sort of non-randomness can arise in multiple ways. A major finding of the work is that the size of a host clade can be an important determinant of its overall infection level. This had been shown in previous work, assuming that the host phylogeny was fixed, but the current paper shows that it extends also to situations where host extinction and speciation takes place at a comparable rate to host shifting. This finding, then, calls into question the natural assumption that a clade of host species that is highly parasite ridden, must have some genetic or ecological characteristic that makes them particularly prone to infection, arguing that the clade size, rather than any characteristic of the clade members, might be the important factor. It will be interesting to see whether this prediction about clade size is borne out with comparative studies. Another feature of the study is that the framework is naturally extendable, to include further processes, such as the influence of parasite presence on extinction or speciation rates. No doubt extensions of this kind will form the basis of important future work. References [1] Engelstädter J and Fortuna NZ. 2018. The dynamics of preferential host switching: host phylogeny as a key predictor of parasite prevalence and distribution. bioRxiv 209254 ver. 5 peer-reviewed by Peer Community In Evolutionary Biology. doi: 10.1101/209254 | The dynamics of preferential host switching: host phylogeny as a key predictor of parasite prevalence and distribution | Jan Engelstaedter & Nicole Fortuna | <p>New parasites commonly arise through host-shifts, where parasites from one host species jump to and become established in a new host species. There is much evidence that the probability of host-shifts decreases with increasing phylogenetic dist... | | Bioinformatics & Computational Biology, Evolutionary Epidemiology, Evolutionary Theory, Macroevolution, Phylogenetics / Phylogenomics, Species interactions | Lucy Weinert | 2017-10-30 02:06:06 | ||
12 Jun 2018

Transgenerational cues about local mate competition affect offspring sex ratios in the spider mite Tetranychus urticaeMaternal effects in sex-ratio adjustmentRecommended by Dries Bonte based on reviews by 2 anonymous reviewersOptimal sex ratios have been topic of extensive studies so far. Fisherian 1:1 proportions of males and females are known to be optimal in most (diploid) organisms, but many deviations from this golden rule are observed. These deviations not only attract a lot of attention from evolutionary biologists but also from population ecologists as they eventually determine long-term population growth. Because sex ratios are tightly linked to fitness, they can be under strong selection or plastic in response to changing demographic conditions. Hamilton [1] pointed out that an equality of the sex ratio breaks down when there is local competition for mates. Competition for mates can be considered as a special case of local resource competition. In short, this theory predicts females to adjust their offspring sex ratio conditional on cues indicating the level of local mate competition that their sons will experience. When cues indicate high levels of LMC mothers should invest more resources in the production of daughters to maximise their fitness, while offspring sex ratios should be closer to 50:50 when cues indicate low levels of LMC. References [1] Hamilton, W. D. (1967). Extraordinary Sex Ratios. Science, 156(3774), 477–488. doi: 10.1126/science.156.3774.477 | Transgenerational cues about local mate competition affect offspring sex ratios in the spider mite Tetranychus urticae | Alison B. Duncan, Cassandra Marinosci, Céline Devaux, Sophie Lefèvre, Sara Magalhães, Joanne Griffin, Adeline Valente, Ophélie Ronce, Isabelle Olivieri | <p style="text-align: justify;">In structured populations, competition between closely related males for mates, termed Local Mate Competition (LMC), is expected to select for female-biased offspring sex ratios. However, the cues underlying sex all... | | Evolutionary Ecology, Life History | Dries Bonte | 2017-12-29 16:10:32 | ||
05 May 2020
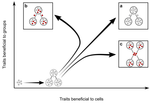
Meta-population structure and the evolutionary transition to multicellularityThe ecology of evolutionary transitions to multicellularityRecommended by Dustin Brisson based on reviews by 2 anonymous reviewersThe evolutionary transition to multicellular life from free-living, single-celled ancestors has occurred independently in multiple lineages [1-5]. This evolutionary transition to cooperative group living can be difficult to explain given the fitness advantages enjoyed by the non-cooperative, single-celled organisms that still numerically dominate life on earth [1,6,7]. Although several hypotheses have been proposed to explain the transition to multicellularity, a common theme is the abatement of the efficacy of natural selection among the single cells during the free-living stage and the promotion of the efficacy of selection among groups of cells during the cooperative stage, an argument reminiscent of those from George Williams’ seminal book [8,9]. The evolution of life cycles appears to be a key step in the transition to multicellularity as it can align fitness advantages of the single-celled 'reproductive' stage with that of the cooperative 'organismal' stage [9-12]. That is, the evolution of life cycles allows natural selection to operate over timescales longer than that of the doubling time of the free-living cells [13]. Despite the importance of this issue, identifying the range of ecological conditions that reduce the importance of natural selection at the single-celled, free-living stage and increase the importance of selection among groups of cooperating cells has not been addressed empirically. References [1] Maynard Smith, J. and Szathmáry, E. (1995). The Major Transitions in Evolution. Oxford, UK: Freeman. p 346. | Meta-population structure and the evolutionary transition to multicellularity | Caroline J Rose, Katrin Hammerschmidt, Yuriy Pichugin and Paul B Rainey | <p>The evolutionary transition to multicellularity has occurred on numerous occasions, but transitions to complex life forms are rare. While the reasons are unclear, relevant factors include the intensity of within- versus between-group selection ... | | Adaptation, Evolutionary Dynamics, Experimental Evolution | Dustin Brisson | 2019-04-04 12:26:36 | ||
06 Mar 2023
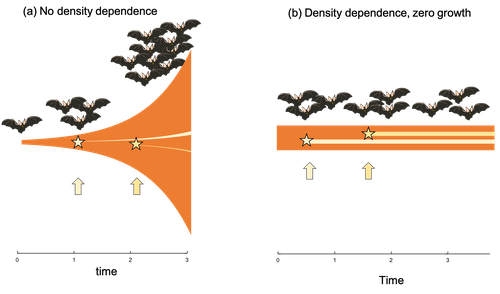
Extrinsic mortality and senescence: a guide for the perplexedGetting old gracefully, and risk of dying before getting there: a new guide to theory on extrinsic mortality and senescenceRecommended by Sinead English and Shinichi Nakagawa based on reviews by Nicole Walasek and 1 anonymous reviewerWhy is there such variation across species and populations in the rate at which individuals show wear and tear as they get older? Several compelling theoretical explanations have been developed on the conditions under which selection allows for or prevents senescence; a notable one being that proposed by George C Williams in 1957 based on the idea of antagonistic pleiotropy (Williams, 1957). One of the testable predictions of this theory is that, in populations where adults experience higher mortality, senescence is expected to be faster. This is one of the most influential predictions of the paper, being intuitive (when individuals are less likely to survive to later age classes, we expect weakened selection on traits that would avoid senescence in these classes), and fitting with ‘live fast, die young’ life history framing. As such, it has been widely incorporated into how we think about the evolution of senescence and has received considerable support in comparative studies across species and populations. However, it would be misleading to sit back at this point and think we have ‘solved’ the problem of understanding variation in senescence, and how this is linked with mortality. It turns out that the Williams 1957 paper is hotly contested by theoreticians: for the past 30 years – with increasing focus in the last 4 years – a growing body of models and opinion pieces have proposed flaws in the paper itself and in how it has been interpreted (Abrams, 1993; André and Rousset, 2020; Day and Abrams, 2020; Moorad et al., 2019). Central to several of these critiques is that explicit consideration of density dependence (not considered in Williams’ original paper) changes the conditions under which his predictions hold. A new preprint by de Vries, Gallipaud and Kokko brings further clarity to such critiques of the original paper (Vries et al., 2023). Beyond just continuing the tradition of critiquing Williams’ prediction, however, de Vries et al. provide a clear guide that is accessible to non-theoreticians about the issues with William’s prediction, and the way in which density dependence and how it operates can change when we expect senescence to occur. Rather than reiterate their points here, we suggest a close reading of the paper itself, along with an excellent overview of the paper in a recent blog by Daniel Nettle (Nettle, 2022). In brief, the paper starts by synthesizing earlier theoretical and empirical studies on the topic and presenting a very simple model to highlight how – in the absence of density dependence – Williams’ prediction does not hold because of the unregulated population growth, which is necessarily higher when there is low mortality. As a result, for a lineage with low mortality, any fitness advantage of placing offspring into the lineage later (i.e. selection for reduced senescence) is exactly cancelled out by the fact that earlier-produced offspring have higher fitness returns. They then present a more complex framework, which incorporates realistic mortality distributions, trade-offs between survival and reproduction, and use a series of 10 scenarios of density dependence (and whether this acts on survival or fecundity, and also whether it depends on a threshold or stochastic, or exerts continuing pressure on the trait) to explore selection on fitness-associated traits with age depending on extrinsic mortality. This then generates a range of results for when the Williams prediction holds, when there is no link between mortality and senescence, and when there is an ‘anti-Williams’ result – i.e., where senescence is slower when there is a high mortality. As has been shown in earlier studies, density dependence and how it operates matters, and Williams’ prediction holds most when density dependence affects juvenile age classes (in their model, when adults are less likely to produce them – i.e. there is density dependence on fecundity; or when there is less recruitment into the adult population due to, for example, competition among juveniles). So, perhaps we are now at a point where we can lay to rest the debate on the issues specifically with Williams’ original paper, and instead consider more broadly the key factors to measure when predicting patterns of senescence, and what is tangible for empiricists to quantify in their studies. Here, de Vries et al. provide some helpful insights both into the limitations of their approach and what modelling should be done moving forward, and into how we can link existing studies (for example comparing senescence among populations with varying predation pressure) to the theoretical predictions. At the heart of the latter is understanding the mechanism of density-dependent regulation – does it affect survival or fecundity, which age classes are most sensitive, and how do key traits depend on density? – and this is often difficult to measure in field studies. And from all this we can learn that even very intuitive and extensively discussed concepts in biology do not always have as firm theoretical underpinnings as assumed, and – as should not be surprising – biology is complex and rather than one clear pattern being predicted, this can depend on a multitude of factors. REFERENCES Abrams PA (1993) Does increased mortality favor the evolution of more rapid senescence? Evolution, 47, 877–887. https://doi.org/10.1111/j.1558-5646.1993.tb01241.x André J-B, Rousset F (2020) Does extrinsic mortality accelerate the pace of life? A bare-bones approach. Evolution and Human Behavior, 41, 486–492. https://doi.org/10.1016/j.evolhumbehav.2020.03.002 Day T, Abrams PA (2020) Density Dependence, Senescence, and Williams’ Hypothesis. Trends in Ecology & Evolution, 35, 300–302. https://doi.org/10.1016/j.tree.2019.11.005 Moorad J, Promislow D, Silvertown J (2019) Evolutionary Ecology of Senescence and a Reassessment of Williams’ ‘Extrinsic Mortality’ Hypothesis. Trends in Ecology & Evolution, 34, 519–530. https://doi.org/10.1016/j.tree.2019.02.006 Nettle AD (2022) Live fast and die young (maybe). https://www.danielnettle.org.uk/2022/02/18/live-fast-and-die-young-maybe/ (accessed 2.27.23). de Vries C, Galipaud M, Kokko H (2023) Extrinsic mortality and senescence: a guide for the perplexed. bioRxiv, 2022.01.27.478060, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.01.27.478060 Williams GC (1957) Pleiotropy, natural selection, and the evolution of senescence. Evolution, 11, 398–411. https://doi.org/10.1111/j.1558-5646.1957.tb02911.x | Extrinsic mortality and senescence: a guide for the perplexed | Charlotte de Vries, Matthias Galipaud, Hanna Kokko | <p style="text-align: justify;">Do environments or species traits that lower the mortality of individuals create selection for delaying senescence? Reading the literature creates an impression that mathematically oriented biologists cannot agree o... | | Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Theory, Life History | Sinead English | 2022-08-26 14:30:16 | ||
22 May 2023
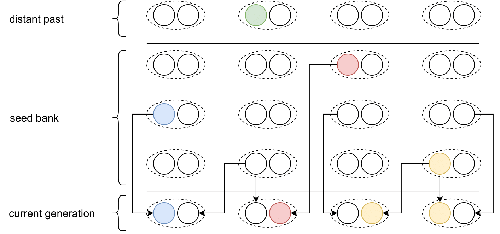
Weak seed banks influence the signature and detectability of selective sweepsNew insights into the dynamics of selective sweeps in seed-banked speciesRecommended by Renaud Vitalis based on reviews by Guillaume Achaz, Jere Koskela, William Shoemaker and Simon Boitard
Many organisms across the Tree of life have the ability to produce seeds, eggs, cysts, or spores, that can remain dormant for several generations before hatching. This widespread adaptive trait in bacteria, fungi, plants and animals, has a significant impact on the ecology, population dynamics and population genetics of species that express it (Evans and Dennehy 2005). In population genetics, and despite the recognition of its evolutionary importance in many empirical studies, few theoretical models have been developed to characterize the evolutionary consequences of this trait on the level and distribution of neutral genetic diversity (see, e.g., Kaj et al. 2001; Vitalis et al. 2004), and also on the dynamics of selected alleles (see, e.g., Živković and Tellier 2018). However, due to the complexity of the interactions between evolutionary forces in the presence of dormancy, the fate of selected mutations in their genomic environment is not yet fully understood, even from the most recently developed models. In a comprehensive article, Korfmann et al. (2023) aim to fill this gap by investigating the effect of germ banking on the probability of (and time to) fixation of beneficial mutations, as well as on the shape of the selective sweep in their vicinity. To this end, Korfmann et al. (2023) developed and released their own forward-in-time simulator of genome-wide data, including neutral and selected polymorphisms, that makes use of Kelleher et al.’s (2018) tree sequence toolkit to keep track of gene genealogies. The originality of Korfmann et al.’s (2023) study is to provide new quantitative results for the effect of dormancy on the time to fixation of positively selected mutations, the shape of the genomic landscape in the vicinity of these mutations, and the temporal dynamics of selective sweeps. Their major finding is the prediction that germ banking creates narrower signatures of sweeps around positively selected sites, which are detectable for increased periods of time (as compared to a standard Wright-Fisher population). The availability of Korfmann et al.’s (2023) code will allow a wider range of parameter values to be explored, to extend their results to the particularities of the biology of many species. However, as they chose to extend the haploid coalescent model of Kaj et al. (2001), further development is needed to confirm the robustness of their results with a more realistic diploid model of seed dormancy. REFERENCES Evans, M. E. K., and J. J. Dennehy (2005) Germ banking: bet-hedging and variable release from egg and seed dormancy. The Quarterly Review of Biology, 80(4): 431-451. https://doi.org/10.1086/498282 Kaj, I., S. Krone, and M. Lascoux (2001) Coalescent theory for seed bank models. Journal of Applied Probability, 38(2): 285-300. https://doi.org/10.1239/jap/996986745 Kelleher, J., K. R. Thornton, J. Ashander, and P. L. Ralph (2018) Efficient pedigree recording for fast population genetics simulation. PLoS Computational Biology, 14(11): e1006581. https://doi.org/10.1371/journal.pcbi.1006581 Korfmann, K., D. Abu Awad, and A. Tellier (2023) Weak seed banks influence the signature and detectability of selective sweeps. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.04.26.489499 Vitalis, R., S. Glémin, and I. Olivieri (2004) When genes go to sleep: the population genetic consequences of seed dormancy and monocarpic perenniality. American Naturalist, 163(2): 295-311. https://doi.org/10.1086/381041 Živković, D., and A. Tellier (2018). All but sleeping? Consequences of soil seed banks on neutral and selective diversity in plant species. Mathematical Modelling in Plant Biology, 195-212. https://doi.org/10.1007/978-3-319-99070-5_10 | Weak seed banks influence the signature and detectability of selective sweeps | Kevin Korfmann, Diala Abu Awad, Aurélien Tellier | <p style="text-align: justify;">Seed banking (or dormancy) is a widespread bet-hedging strategy, generating a form of population overlap, which decreases the magnitude of genetic drift. The methodological complexity of integrating this trait impli... | | Adaptation, Bioinformatics & Computational Biology, Evolutionary Applications, Evolutionary Ecology, Genome Evolution, Life History, Population Genetics / Genomics | Renaud Vitalis | 2022-05-23 13:01:57 | ||
18 Aug 2020
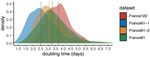
Early phylodynamics analysis of the COVID-19 epidemics in FranceSARS-Cov-2 genome sequence analysis suggests rapid spread followed by epidemic slowdown in FranceRecommended by B. Jesse Shapiro based on reviews by Luca Ferretti and 2 anonymous reviewersSequencing and analyzing SARS-Cov-2 genomes in nearly real time has the potential to quickly confirm (and inform) our knowledge of, and response to, the current pandemic [1,2]. In this manuscript [3], Danesh and colleagues use the earliest set of available SARS-Cov-2 genome sequences available from France to make inferences about the timing of the major epidemic wave, the duration of infections, and the efficacy of lockdown measures. Their phylodynamic estimates -- based on fitting genomic data to molecular clock and transmission models -- are reassuringly close to estimates based on 'traditional' epidemiological methods: the French epidemic likely began in mid-January or early February 2020, and spread relatively rapidly (doubling every 3-5 days), with people remaining infectious for a median of 5 days [4,5]. These transmission parameters are broadly in line with estimates from China [6,7], but are currently unknown in France (in the absence of contact tracing data). By estimating the temporal reproductive number (Rt), the authors detected a slowing down of the epidemic in the most recent period of the study, after mid-March, supporting the efficacy of lockdown measures. References [1] Grubaugh, N. D., Ladner, J. T., Lemey, P., Pybus, O. G., Rambaut, A., Holmes, E. C., & Andersen, K. G. (2019). Tracking virus outbreaks in the twenty-first century. Nature microbiology, 4(1), 10-19. doi: 10.1038/s41564-018-0296-2 | Early phylodynamics analysis of the COVID-19 epidemics in France | Gonché Danesh, Baptiste Elie,Yannis Michalakis, Mircea T. Sofonea, Antonin Bal, Sylvie Behillil, Grégory Destras, David Boutolleau, Sonia Burrel, Anne-Geneviève Marcelin, Jean-Christophe Plantier, Vincent Thibault, Etienne Simon-Loriere, Sylvie va... | <p>France was one of the first countries to be reached by the COVID-19 pandemic. Here, we analyse 196 SARS-Cov-2 genomes collected between Jan 24 and Mar 24 2020, and perform a phylodynamics analysis. In particular, we analyse the doubling time, r... | | Evolutionary Epidemiology, Molecular Evolution, Phylogenetics / Phylogenomics | B. Jesse Shapiro | 2020-06-04 13:13:57 |




