
Latest recommendations

| Id | Title | Authors | Abstract▼ | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
06 Oct 2017

Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebratesCombining molecular information on chromatin organisation with eQTLs and evolutionary conservation provides strong candidates for the evolution of gene regulation in mammalian brainsRecommended by Marc Robinson-Rechavi based on reviews by Marc Robinson-Rechavi and Charles DankoIn this manuscript [1], Francisco J. Novo proposes candidate non-coding genomic elements regulating neurodevelopmental genes. What is very nice about this study is the way in which public molecular data, including physical interaction data, is used to leverage recent advances in our understanding to molecular mechanisms of gene regulation in an evolutionary context. More specifically, evolutionarily conserved non coding sequences are combined with enhancers from the FANTOM5 project, DNAse hypersensitive sites, chromatin segmentation, ChIP-seq of transcription factors and of p300, gene expression and eQTLs from GTEx, and physical interactions from several Hi-C datasets. The candidate regulatory regions thus identified are linked to candidate regulated genes, and the author shows their potential implication in brain development. While the results are focused on a small number of genes, this allows to verify features of these candidates in great detail. This study shows how functional genomics is increasingly allowing us to fulfill the promises of Evo-Devo: understanding the molecular mechanisms of conservation and differences in morphology. References [1] Novo, FJ. 2017. Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebrates. bioRxiv, 150482, ver. 4 of Sept 29th, 2017. doi: 10.1101/150482 | Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebrates | Francisco J. Novo | <p>Many non-coding regulatory elements conserved in vertebrates regulate the expression of genes involved in development and play an important role in the evolution of morphology through the rewiring of developmental gene networks. Available biolo... | | Genome Evolution | Marc Robinson-Rechavi | Marc Robinson-Rechavi, Charles Danko | 2017-06-29 08:55:41 | |
12 Nov 2020
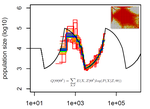
Limits and Convergence properties of the Sequentially Markovian CoalescentReview and Assessment of Performance of Genomic Inference Methods based on the Sequentially Markovian CoalescentRecommended by Stephan Schiffels based on reviews by 3 anonymous reviewers based on reviews by 3 anonymous reviewers
The human genome not only encodes for biological functions and for what makes us human, it also encodes the population history of our ancestors. Changes in past population sizes, for example, affect the distribution of times to the most recent common ancestor (tMRCA) of genomic segments, which in turn can be inferred by sophisticated modelling along the genome. References [1] Li, H., and Durbin, R. (2011). Inference of human population history from individual whole-genome sequences. Nature, 475(7357), 493-496. doi: https://doi.org/10.1038/nature10231 | Limits and Convergence properties of the Sequentially Markovian Coalescent | Thibaut Sellinger, Diala Abu Awad, Aurélien Tellier | <p>Many methods based on the Sequentially Markovian Coalescent (SMC) have been and are being developed. These methods make use of genome sequence data to uncover population demographic history. More recently, new methods have extended the original... | | Population Genetics / Genomics | Stephan Schiffels | Anonymous | 2020-07-25 10:54:48 | |
06 Sep 2022

Masculinization of the X-chromosome in aphid soma and gonadsSex-biased gene expression is not tissue-specific in Pea AphidsRecommended by Charles Baer and Tanja Schwander based on reviews by Ann Kathrin Huylmans and 1 anonymous reviewerSexual antagonism (SA), wherein the fitness interests of the sexes do not align, is inherent to organisms with two (or more) sexes. SA leads to intra-locus sexual conflict, where an allele that confers higher fitness in one sex reduces fitness in the other [1, 2]. This situation leads to what has been referred to as "gender load", resulting from the segregation of SA alleles in the population. Gender load can be reduced by the evolution of sex-specific (or sex-biased) gene expression. A specific prediction is that gene-duplication can lead to sub- or neo-functionalization, in which case the two duplicates partition the function in the different sexes. The conditions for invasion by a SA allele differ between sex-chromosomes and autosomes, leading to the prediction that (in XY or XO systems) the X should accumulate recessive male-favored alleles and dominant female-favored alleles; similar considerations apply in ZW systems ([3, but see 4]. Aphids present an interesting special case, for several reasons: they have XO sex-determination, and three distinct reproductive morphs (sexual females, parthenogenetic females, and males). Previous theoretical work by the lead author predict that the X should be optimized for male function, which was borne out by whole-animal transcriptome analysis [5]. Here [6], the authors extend that work to investigate “tissue”-specific (heads, legs and gonads), sex-specific gene expression. They argue that, if intra-locus SA is the primary driver of sex-biased gene expression, it should be generally true in all tissues. They set up as an alternative the possibility that sex-biased gene expression could also be driven by dosage compensation. They cite references supporting their argument that "dosage compensation (could be) stronger in the brain", although the underlying motivation for that argument appears to be based on empirical evidence rather than theoretical predictions. At any rate, the results are clear: all tissues investigated show masculinization of the X. Further, X-linked copies of gene duplicates were more frequently male-biased than duplicated autosomal genes or X-linked single-copy genes. To sum up, this is a nice empirical study with clearly interpretable (and interpreted) results, the most obvious of which is the greater sex-biased expression in sexually-dimorphic tissues. Unfortunately, as the authors emphasize, there is no general theory by which SA, variable dosage-compensation, and meiotic sex chromosome inactivation can be integrated in a predictive framework. It is to be hoped that empirical studies such as this one will motivate deeper and more general theoretical investigations. References [1] Rice WR, Chippindale AK (2001) Intersexual ontogenetic conflict. Journal of Evolutionary Biology 14: 685-693. https://doi.org/10.1046/j.1420-9101.2001.00319.x [2] Bonduriansky R, Chenoweth SF (2009) Intralocus sexual conflict. Trends Ecol Evol 24: 280-288. https://doi.org/10.1016/j.tree.2008.12.005 [3] Rice WR. (1984) Sex chromosomes and the evolution of sexual dimorphism. Evolution 38: 735-742. https://doi.org/10.1086/595754 [4] Fry JD (2010) The genomic location of sexually antagonistic variation: some cautionary comments. Evolution 64: 1510-1516. https://doi.org/10.1111%2Fj.1558-5646.2009.00898.x [5] Jaquiéry J, Rispe C, Roze D, Legeai F, Le Trionnaire G, Stoeckel S, et al. (2013) Masculinization of the X Chromosome in the Pea Aphid. PLoS Genetics 9. https://doi.org/10.1371/journal.pgen.1003690 [6] Jaquiéry J, Simon J-C, Robin S, Richard G, Peccoud J, Boulain H, Legeai F, Tanguy S, Prunier-Leterme N, Le Trionnaire G (2022) Masculinization of the X-chromosome in aphid soma and gonads. bioRxiv, 2021.08.13.453080, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.08.13.453080 | Masculinization of the X-chromosome in aphid soma and gonads | Julie Jaquiery, Jean-Christophe Simon, Stephanie Robin, Gautier Richard, Jean Peccoud, Helene Boulain, Fabrice Legeai, Sylvie Tanguy, Nathalie Prunier-Leterme, Gael Letrionnaire | <p>Males and females share essentially the same genome but differ in their optimal values for many phenotypic traits, which can result in intra-locus conflict between the sexes. Aphids display XX/X0 sex chromosomes and combine unusual X chromosome... | | Genetic conflicts, Genome Evolution, Reproduction and Sex | Charles Baer | 2021-08-16 08:56:08 | ||
15 Feb 2019

Architectural traits constrain the evolution of unisexual flowers and sexual segregation within inflorescences: an interspecific approachSometimes, sex is in the headRecommended by Juan Arroyo based on reviews by 3 anonymous reviewers
Plants display an amazing diversity of reproductive strategies with and without sex. This diversity is particularly remarkable in flowering plants, as highlighted by Charles Darwin, who wrote several botanical books scrutinizing plant reproduction. One particularly influential work concerned floral variation [1]. Darwin recognized that flowers may present different forms within a single population, with or without sex specialization. The number of species concerned is small, but they display recurrent patterns, which made it possible for Darwin to invoke natural and sexual selection to explain them. Most of early evolutionary theory on the evolution of reproductive strategies was developed in the first half of the 20th century and was based on animals. However, the pioneering work by David Lloyd from the 1970s onwards excited interest in the diversity of plant sexual strategies as models for testing adaptive hypotheses and predicting reproductive outcomes [2]. The sex specialization of individual flowers and plants has since become one of the favorite topics of evolutionary biologists. However, attention has focused mostly on cases related to sex differentiation (dioecy and associated conditions [3]). Separate unisexual flower types on the same plant (monoecy and related cases, rendering the plant functionally hermaphroditic) have been much less studied, apart from their possible role in the evolution of dioecy [4] or their association with particular modes of pollination [5]. References [1] Darwin, C. (1877). The different forms of flowers on plants of the same species. John Murray. | Architectural traits constrain the evolution of unisexual flowers and sexual segregation within inflorescences: an interspecific approach | Rubén Torices, Ana Afonso, Arne A. Anderberg, José M. Gómez and Marcos Méndez | <p>Male and female unisexual flowers have repeatedly evolved from the ancestral bisexual flowers in different lineages of flowering plants. This sex specialization in different flowers often occurs within inflorescences. We hypothesize that inflor... | | Evolutionary Ecology, Morphological Evolution, Phenotypic Plasticity, Reproduction and Sex, Sexual Selection | Juan Arroyo | Jana Vamosi, Marcial Escudero, Anonymous | 2018-06-27 10:49:52 | |
23 Jan 2023
The genetic architecture of local adaptation in a clineEnvironmental and fitness landscapes matter for the genetic basis of local adaptationRecommended by Charles Mullon based on reviews by 2 anonymous reviewers
Natural landscapes are often composite, with spatial variation in environmental factors being the norm rather than exception. Adaptation to such variation is a major driver of diversity at all levels of biological organization, from genes to phenotypes, species and ultimately ecosystems. While natural selection favours traits that show a better fit to local conditions, the genomic response to such selection is not necessarily straightforward. This is because many quantitative traits are complex and the product of many loci, each with a small to moderate phenotypic contribution. Adapting to environmental challenges that occur in narrow ranges may thus prove difficult as each individual locus is easily swamped by alleles favoured across the rest of the population range. To better understand whether and how evolution overcomes such a hurdle, Laroche and Lenormand [1] combine quantitative genetics and population genetic modelling to track genomic changes that underpin a trait whose fitness optimum differs between a certain spatial range, referred to as a “pocket”, and the rest of the habitat. As it turns out from their analysis, one critical and probably underappreciated factor in determining the type of genetic architecture that evolves is how fitness declines away from phenotypic optima. One classical and popular model of fitness landscape that relates trait value to reproductive success is Gaussian, whereby small trait variations away from the optimum result in even smaller variations in fitness. This facilitates local adaptation via the invasion of alleles of small effects as carriers inside the pocket show a better fit while those outside the pocket only suffer a weak fitness cost. By contrast, when the fitness landscape is more peaked around the optimum, for instance where the decline is linear, adaptation through weak effect alleles is less likely, requiring larger pockets that are less easily swamped by alleles selected in the rest of the range. In addition to mathematically investigating the initial emergence of local adaptation, Laroche and Lenormand use computer simulations to look at its long-term maintenance. In principle, selection should favour a genetic architecture that consolidates the phenotype and increases its heritability, for instance by grouping several alleles of large effects close to one another on a chromosome to avoid being broken down by meiotic recombination. Whether or not this occurs also depends on the fitness landscape. When the landscape is Gaussian, the genetic architecture of the trait eventually consists of tightly linked alleles of large effects. The replacement of small effects by large effects loci is here again promoted by the slow fitness decline around the optimum. This is because any shift in architecture in an adapted population requires initially crossing a fitness valley. With a Gaussian landscape, this valley is shallow enough to be crossed, facilitated by a bit of genetic drift. By contrast, when fitness declines linearly around the optimum, genetic architecture is much less evolutionarily labile as any architecture change initially entails a fitness cost that is too high to bear. Overall, Laroche and Lenormand provide a careful and thought-provoking analysis of a classical problem in population genetics. In addition to questioning some longstanding modelling assumptions, their results may help understand why differentiated populations are sometimes characterized by “genomic islands” of divergence, and sometimes not. References [1] Laroche F, Lenormand T (2022) The genetic architecture of local adaptation in a cline. bioRxiv, 2022.06.30.498280, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.06.30.498280 | The genetic architecture of local adaptation in a cline | Fabien Laroche, Thomas Lenormand | <p>Local adaptation is pervasive. It occurs whenever selection favors different phenotypes in different environments, provided that there is genetic variation for the corresponding traits and that the effect of selection is greater than the effect... | | Adaptation, Evolutionary Theory, Genome Evolution, Molecular Evolution, Population Genetics / Genomics, Quantitative Genetics | Charles Mullon | 2022-07-07 08:46:47 | ||
08 Oct 2019

Strong habitat and weak genetic effects shape the lifetime reproductive success in a wild clownfish populationHabitat variation of wild clownfish population shapes selfrecruitment more than genetic effectsRecommended by Philip Munday based on reviews by Juan Diego Gaitan-Espitia and Loeske KruukEstimating the genetic and environmental components of variation in reproductive success is crucial to understanding the adaptive potential of populations to environmental change. To date, the heritability of lifetime reproductive success (fitness) has been estimated in a handful of wild animal population, mostly in mammals and birds, but has never been estimated for a marine species. The primary reason that such estimates are lacking in marine species is that most marine organisms have a dispersive larval phase, making it extraordinarily difficult to track the fate of offspring from one generation to the next. References [1] Salles, O. C., Almany, G. R., Berumen, M.L., Jones, G. P., Saenz-Agudelo, P., Srinivasan, M., Thorrold, S. R., Pujol, B., Planes, S. (2019). Strong habitat and weak genetic effects shape the lifetime reproductive success in a wild clownfish population. Zenodo, 3476529, ver. 3 peer-reviewed and recommended by Peer Community In Evolutionary Biology. doi: 10.5281/zenodo.3476529 | Strong habitat and weak genetic effects shape the lifetime reproductive success in a wild clownfish population | Océane C. Salles, Glenn R. Almany, Michael L. Berumen, Geoffrey P. Jones, Pablo Saenz-Agudelo, Maya Srinivasan, Simon Thorrold, Benoit Pujol, Serge Planes | <p>Lifetime reproductive success (LRS), the number of offspring an individual contributes to the next generation, is of fundamental importance in ecology and evolutionary biology. LRS may be influenced by environmental, maternal and additive genet... | | Adaptation, Evolutionary Ecology, Life History, Quantitative Genetics | Philip Munday | 2018-10-01 09:00:53 | ||
05 Apr 2024
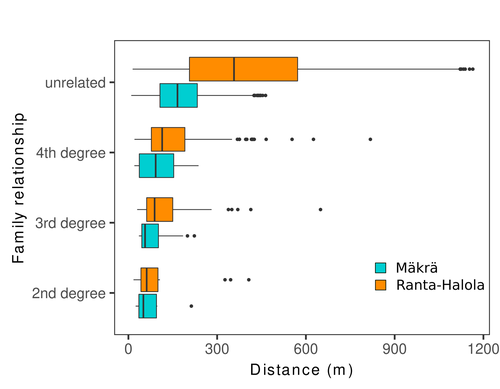
Does the seed fall far from the tree? Weak fine scale genetic structure in a continuous Scots pine populationWeak spatial genetic structure in a large continuous Scots pine population – implications for conservation and breedingRecommended by Myriam Heuertz based on reviews by Joachim Mergeay, Jean-Baptiste Ledoux and Roberta Loh
Spatial genetic structure, i.e. the non-random spatial distribution of genotypes, arises in populations because of different processes including spatially limited dispersal and selection. Knowledge on the spatial genetic structure of plant populations is important to assess biological parameters such as gene dispersal distances and the potential for local adaptations, as well as for applications in conservation management and breeding. In their work, Niskanen and colleagues demonstrate a multifaceted approach to characterise the spatial genetic structure in two replicate sites of a continuously distributed Scots pine population in South-Eastern Finland. They mapped and assessed the ages of 469 naturally regenerated adults and genotyped them using a SNP array which resulted in 157 325 filtered polymorphic SNPs. Their dataset is remarkably powerful because of the large numbers of both individuals and SNPs genotyped. This made it possible to characterise precisely the decay of genetic relatedness between individuals with spatial distance despite the extensive dispersal capacity of Scots pine through pollen, and ensuing expectations of an almost panmictic population. The authors’ data analysis was particularly thorough. They demonstrated that two metrics of pairwise relatedness, the genomic relationship matrix (GRM, Yang et al. 2011) and the kinship coefficient (Loiselle et al. 1995) were strongly correlated and produced very similar inference of family relationships: >99% of pairs of individuals were unrelated, and the remainder exhibited 2nd (e.g., half-siblings) to 4th degree relatedness. Pairwise relatedness decayed with spatial distance which resulted in extremely weak but statistically significant spatial genetic structure in both sites, quantified as Sp=0.0005 and Sp=0.0008. These estimates are at least an order of magnitude lower than estimates in the literature obtained in more fragmented populations of the same species or in other conifers. Estimates of the neighbourhood size, the effective number of potentially mating individuals belonging to a within-population neighbourhood (Wright 1946), were relatively large with Nb=1680-3210 despite relatively short gene dispersal distances, σg = 36.5–71.3m, which illustrates the high effective density of the population. The authors showed the implications of their findings for selection. The capacity for local adaptation depends on dispersal distances and the strength of the selection coefficient. In the study population, the authors inferred that local adaptation can only occur if environmental heterogeneity occurs over a distance larger than approximately one kilometre (or larger, if considering long-distance dispersal). Interestingly, in Scots pine, no local adaptation has been described on similar geographic scales, in contrast to some other European or Mediterranean conifers (Scotti et al. 2023). The authors’ results are relevant for the management of conservation and breeding. They showed that related individuals occurred within sites only and that they shared a higher number of rare alleles than unrelated ones. Since rare alleles are enriched in new and recessive deleterious variants, selecting related individuals could have negative consequences in breeding programmes. The authors also showed, in their response to reviewers, that their powerful dataset was not suitable to obtain a robust estimate of effective population size, Ne, based on the linkage disequilibrium method (Do et al. 2014). This illustrated that the estimation of Ne used for genetic indicators supported in international conservation policy (Hoban et al. 2020, CBD 2022) remains challenging in large and continuous populations (see also Santo-del-Blanco et al. 2023, Gargiulo et al. 2024). ReferencesCBD (2022) Kunming-Montreal Global Biodiversity Framework. https://www.cbd.int/doc/decisions/cop-15/cop-15-dec-04-en.pdf Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014). NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne ) from genetic data. Molecular Ecology Resources 14: 209–214. https://doi.org/10.1111/1755-0998.12157 Gargiulo R, Decroocq V, González-Martínez SC, Paz-Vinas I, Aury JM, Kupin IL, Plomion C, Schmitt S, Scotti I, Heuertz M (2024) Estimation of contemporary effective population size in plant populations: limitations of genomic datasets. Evolutionary Applications, in press, https://doi.org/10.1101/2023.07.18.549323 Hoban S, Bruford M, D’Urban Jackson J, Lopes-Fernandes M, Heuertz M, Hohenlohe PA, Paz-Vinas I, et al. (2020) Genetic diversity targets and indicators in the CBD post-2020 Global Biodiversity Framework must be improved. Biological Conservation 248: 108654. https://doi.org/10.1016/j.biocon.2020.108654 Loiselle BA, Sork VL, Nason J & Graham C (1995) Spatial genetic structure of a tropical understorey shrub, Psychotria officinalis (Rubiaceae). American Journal of Botany 82: 1420–1425. https://doi.org/10.1002/j.1537-2197.1995.tb12679.x Santos-del-Blanco L, Olsson S, Budde KB, Grivet D, González-Martínez SC, Alía R, Robledo-Arnuncio JJ (2022). On the feasibility of estimating contemporary effective population size (Ne) for genetic conservation and monitoring of forest trees. Biological Conservation 273: 109704. https://doi.org/10.1016/j.biocon.2022.109704 Scotti I, Lalagüe H, Oddou-Muratorio S, Scotti-Saintagne C, Ruiz Daniels R, Grivet D, et al. (2023) Common microgeographical selection patterns revealed in four European conifers. Molecular Ecology 32: 393-411. https://doi.org/10.1111/mec.16750 Wright S (1946) Isolation by distance under diverse systems of mating. Genetics 31: 39–59. https://doi.org/10.1093/genetics/31.1.39 Yang J, Lee SH, Goddard ME & Visscher PM (2011) GCTA: a tool for genome-wide complex trait analysis. The American Journal of Human Genetics 88: 76–82. https://www.cell.com/ajhg/pdf/S0002-9297(10)00598-7.pdf | Does the seed fall far from the tree? Weak fine scale genetic structure in a continuous Scots pine population | Alina K. Niskanen, Sonja T. Kujala, Katri Kärkkäinen, Outi Savolainen, Tanja Pyhäjärvi | <p>Knowledge of fine-scale spatial genetic structure, i.e., the distribution of genetic diversity at short distances, is important in evolutionary research and in practical applications such as conservation and breeding programs. In trees, related... | | Adaptation, Evolutionary Applications, Population Genetics / Genomics | Myriam Heuertz | Joachim Mergeay | 2023-06-27 21:57:28 | |
22 Oct 2019

Geographic variation in adult and embryonic desiccation tolerance in a terrestrial-breeding frogTough as old boots: amphibians from drier habitats are more resistant to desiccation, but less flexible at exploiting wet conditionsRecommended by Ben Phillips based on reviews by Juan Diego Gaitan-Espitia, Jennifer Nicole Lohr and 1 anonymous reviewerSpecies everywhere are facing rapid climatic change, and we are increasingly asking whether populations will adapt, shift, or perish [1]. There is a growing realisation that, despite limited within-population genetic variation, many species exhibit substantial geographic variation in climate-relevant traits. This geographic variation might play an important role in facilitating adaptation to climate change [2,3]. References [1] Hoffmann, A. A., and Sgrò, C. M. (2011). Climate change and evolutionary adaptation. Nature, 470(7335), 479–485. doi: 10.1038/nature09670 | Geographic variation in adult and embryonic desiccation tolerance in a terrestrial-breeding frog | Rudin-Bitterli, T, Evans, J. P. and Mitchell, N. J. | <p>Intra-specific variation in the ability of individuals to tolerate environmental perturbations is often neglected when considering the impacts of climate change. Yet this information is potentially crucial for mitigating any deleterious effects... | | Adaptation, Evolutionary Applications, Evolutionary Ecology | Ben Phillips | 2018-05-07 03:35:08 | ||
17 Feb 2020
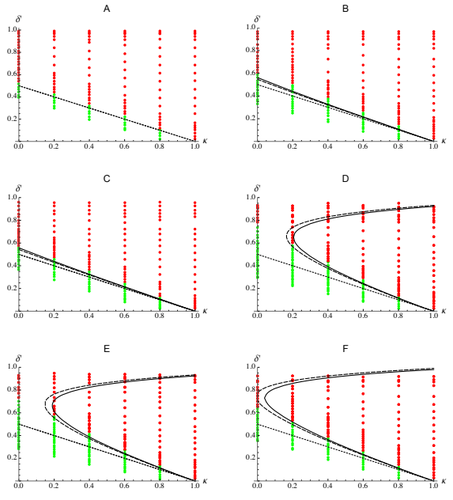
Epistasis, inbreeding depression and the evolution of self-fertilizationEpistasis and the evolution of selfingRecommended by Sylvain Gandon based on reviews by Nick Barton and 1 anonymous reviewerThe evolution of selfing results from a balance between multiple evolutionary forces. Selfing provides an "automatic advantage" due to the higher efficiency of selfers to transmit their genes via selfed and outcrossed offspring. Selfed offspring, however, may suffer from inbreeding depression. In principle the ultimate evolutionary outcome is easy to predict from the relative magnitude of these two evolutionary forces [1,2]. Yet, several studies explicitly taking into account the genetic architecture of inbreeding depression noted that these predictions are often too restrictive because selfing can evolve in a broader range of conditions [3,4]. References [1] Holsinger, K. E., Feldman, M. W., and Christiansen, F. B. (1984). The evolution of self-fertilization in plants: a population genetic model. The American Naturalist, 124(3), 446-453. doi: 10.1086/284287 | Epistasis, inbreeding depression and the evolution of self-fertilization | Diala Abu Awad and Denis Roze | <p>Inbreeding depression resulting from partially recessive deleterious alleles is thought to be the main genetic factor preventing self-fertilizing mutants from spreading in outcrossing hermaphroditic populations. However, deleterious alleles may... | | Evolutionary Theory, Quantitative Genetics, Reproduction and Sex | Sylvain Gandon | 2019-10-18 09:29:41 | ||
25 Mar 2019

The joint evolution of lifespan and self-fertilisationEvolution of selfing & lifespan 2.0Recommended by Thomas Bataillon based on reviews by 2 anonymous reviewersFlowering plants display a staggering diversity of both mating systems and life histories, ranging from almost exclusively selfers to obligate outcrossers, very short-lived annual herbs to super long lived trees. One pervasive pattern that has attracted considerable attention is the tight correlation that is found between mating systems and lifespan [1]. Until recently, theoretical explanations for these patterns have relied on static models exploring the consequences of several non-mutually exclusive important process: levels of inbreeding depression and ability to successfully were center stage. This make sense: successful colonization after long‐distance dispersal is far more likely to happen for self‐compatible than for self‐incompatible individuals in a sexually reproducing species. Furthermore, inbreeding depression (essentially a genetically driven phenomenon) and reproductive insurance are expected to shape the evolution of both mating system and lifespan. References | The joint evolution of lifespan and self-fertilisation | Thomas Lesaffre, Sylvain Billiard | <p>In Angiosperms, there exists a strong association between mating system and lifespan. Most self-fertilising species are short-lived and most predominant or obligate outcrossers are long-lived. This association is generally explained by the infl... | | Evolutionary Theory, Life History, Reproduction and Sex | Thomas Bataillon | 2018-09-19 10:03:51 |




