
Latest recommendations

| Id | Title | Authors | Abstract | Picture | Thematic fields | Recommender▼ | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
06 Oct 2017

Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebratesCombining molecular information on chromatin organisation with eQTLs and evolutionary conservation provides strong candidates for the evolution of gene regulation in mammalian brainsRecommended by Marc Robinson-Rechavi based on reviews by Marc Robinson-Rechavi and Charles DankoIn this manuscript [1], Francisco J. Novo proposes candidate non-coding genomic elements regulating neurodevelopmental genes. What is very nice about this study is the way in which public molecular data, including physical interaction data, is used to leverage recent advances in our understanding to molecular mechanisms of gene regulation in an evolutionary context. More specifically, evolutionarily conserved non coding sequences are combined with enhancers from the FANTOM5 project, DNAse hypersensitive sites, chromatin segmentation, ChIP-seq of transcription factors and of p300, gene expression and eQTLs from GTEx, and physical interactions from several Hi-C datasets. The candidate regulatory regions thus identified are linked to candidate regulated genes, and the author shows their potential implication in brain development. While the results are focused on a small number of genes, this allows to verify features of these candidates in great detail. This study shows how functional genomics is increasingly allowing us to fulfill the promises of Evo-Devo: understanding the molecular mechanisms of conservation and differences in morphology. References [1] Novo, FJ. 2017. Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebrates. bioRxiv, 150482, ver. 4 of Sept 29th, 2017. doi: 10.1101/150482 | Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebrates | Francisco J. Novo | <p>Many non-coding regulatory elements conserved in vertebrates regulate the expression of genes involved in development and play an important role in the evolution of morphology through the rewiring of developmental gene networks. Available biolo... | | Genome Evolution | Marc Robinson-Rechavi | Marc Robinson-Rechavi, Charles Danko | 2017-06-29 08:55:41 | |
08 Jan 2024
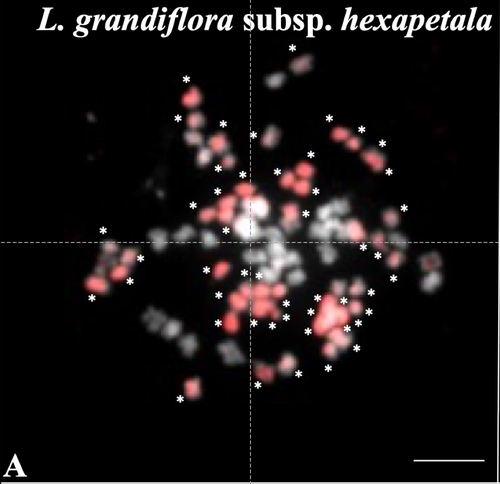
Genomic relationships among diploid and polyploid species of the genus Ludwigia L. section Jussiaea using a combination of molecular cytogenetic, morphological, and crossing investigationsDeciphering the genomic composition of tetraploid, hexaploid and decaploid Ludwigia L. species (section Jussiaea)Recommended by Malika AINOUCHE based on reviews by Alex BAUMEL and Karol MARHOLDPolyploidy, which results in the presence of more than two sets of homologous chromosomes represents a major feature of plant genomes that have undergone successive rounds of duplication followed by more or less rapid diploidization during their evolutionary history. Polyploid complexes containing diploid and derived polyploid taxa are excellent model systems for understanding the short-term consequences of whole genome duplication, and have been particularly well-explored in evolutionary ecology (Ramsey and Ramsey 2014, Rice et al. 2019). Many polyploids (especially when resulting from interspecific hybridization, i.e. allopolyploids) are successful invaders (te Beest et al. 2012) as a result of rapid genome dynamics, functional novelty, and trait evolution. The origin (parental legacy) and modes of formation of polyploids have a critical impact on the subsequent polyploid evolution. Thus, elucidation of the genomic composition of polyploids is fundamental to understanding trait evolution, and such knowledge is still lacking for many invasive species. Genus Ludwigia is characterized by a complex taxonomy, with an underexplored evolutionary history. Species from section Jussieae form a polyploid complex with diploids, tetraploids, hexaploids, and decaploids that are notorious invaders in freshwater and riparian ecosystems (Thouvenot et al.2013). Molecular phylogeny of the genus based on nuclear and chloroplast sequences (Liu et al. 2027) suggested some relationships between diploid and polyploid species, without fully resolving the question of the parentage of the polyploids. In their study, Barloy et al. (2023) have used a combination of molecular cytogenetics (Genomic In situ Hybridization), morphology and experimental crosses to elucidate the genomic compositions of the polyploid species, and show that the examined polyploids are of hybrid origin (allopolyploids). The tetraploid L. stolonifera derives from the diploids L. peploides subsp. montevidensis (AA genome) and L. helminthorhiza (BB genome). The tetraploid L. ascendens also share the BB genome combined with an undetermined different genome. The hexaploid L. grandiflora subsp. grandiflora has inherited the diploid AA genome combined with additional unidentified genomes. The decaploid L. grandiflora subsp. hexapetala has inherited the tetraploid L. stolonifera and the hexaploid L. grandiflora subsp. hexapetala genomes. As the authors point out, further work is needed, including additional related diploid (e.g. other subspecies of L. peploides) or tetraploid (L. hookeri and L. peduncularis) taxa that remain to be investigated, to address the nature of the undetermined parental genomes mentioned above. The presented work (Barloy et al. 2023) provides significant knowledge of this poorly investigated group with regard to genomic information and polyploid origin, and opens perspectives for future studies. The authors also detect additional diagnostic morphological traits of interest for in-situ discrimination of the taxa when monitoring invasive populations. References Barloy D., Portillo-Lemus L., Krueger-Hadfield S.A., Huteau V., Coriton O. (2024). Genomic relationships among diploid and polyploid species of the genus Ludwigia L. section Jussiaea using a combination of molecular cytogenetic, morphological, and crossing investigations. BioRxiv, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology https://doi.org/10.1101/2023.01.02.522458 te Beest M., Le Roux J.J., Richardson D.M., Brysting A.K., Suda J., Kubešová M., Pyšek P. (2012). The more the better? The role of polyploidy in facilitating plant invasions. Annals of Botany, Volume 109, Issue 1 Pages 19–45, https://doi.org/10.1093/aob/mcr277 Ramsey J. and Ramsey T. S. (2014). Ecological studies of polyploidy in the 100 years following its discovery Phil. Trans. R. Soc. B369 1–20 https://doi.org/10.1098/rstb.2013.0352 Rice, A., Šmarda, P., Novosolov, M. et al. (2019). The global biogeography of polyploid plants. Nat Ecol Evol 3, 265–273. https://doi.org/10.1038/s41559-018-0787-9 Thouvenot L, Haury J, Thiebaut G. (2013). A success story: Water primroses, aquatic plant pests. Aquat. Conserv. Mar. Freshw. Ecosyst. 23:790–803 https://doi.org/10.1002/aqc.2387 | Genomic relationships among diploid and polyploid species of the genus *Ludwigia* L. section *Jussiaea* using a combination of molecular cytogenetic, morphological, and crossing investigations | D. Barloy, L. Portillo - Lemus, S. A. Krueger-Hadfield, V. Huteau, O. Coriton | <p>ABSTRACTThe genus Ludwigia L. sectionJussiaeais composed of a polyploid species complex with 2x, 4x, 6x and 10x ploidy levels, suggesting possible hybrid origins. The aim of the present study is to understand the genomic relationships among dip... | | Hybridization / Introgression, Phylogenetics / Phylogenomics | Malika AINOUCHE | 2023-01-11 13:47:18 | ||
20 Dec 2016

POSTPRINT
Experimental Evolution of Gene Expression and Plasticity in Alternative Selective RegimesGenetic adaptation counters phenotypic plasticity in experimental evolutionRecommended by Luis-Miguel Chevin and Stephanie BedhommeHow do phenotypic plasticity and adaptive evolution interact in a novel or changing environment? Does evolution by natural selection generally reinforce initially plastic phenotypic responses, or does it instead oppose them? And to what extent does evolution of a trait involve evolution of its plasticity? These questions have lied at the heart of research on phenotypic evolution in heterogeneous environments ever since it was realized that the environment is likely to affect the expression of many (perhaps most) characters of an individual. Importantly, this broad definition of phenotypic plasticity as change in the average phenotype of a given genotype in response to its environment of development (or expression) does not involve any statement about the adaptiveness of the plastic changes. Theory on the evolution of plasticity has devoted much effort to understanding how reaction norm should evolve under different regimes of environmental change in space and time, and depending on genetic constraints on reaction norm shapes. However on an empirically ground, the questions above have mostly been addressed for individual traits, often chosen a priori for their likeliness to exhibit adaptive plasticity, and we still lack more systematic answers. These can be provided by so-called ‘phenomic’ approaches, where a large number of traits are tracked without prior information on their biological or ecological function. A problem is that the number of phenotypic characters that can be measured in an organism is virtually infinite (and to some extent arbitrary), and that scaling issues makes it difficult to compare different sets of traits. Gene-expression levels offer a partial solution to this dilemma, as they can be considered as a very large number of traits (one per typed gene) that can be measured easily and uniformly (fold change in the number of reads in RNAseq). As for any traits, expression levels of different genes may be genetically correlated, to an extent that depends on their regulation mechanism: cis-regulatory sequences that only affect expression of neighboring genes are likely to cause independent gene expression, while more systematic modifiers of expression (e.g. trans-regulators such as transcription factors) may cause correlated genetic responses of the expression of many genes. Huang and Agrawal [1] have studied plasticity and evolution of gene expression level in young larvae of populations of Drosophila melanogaster that have evolved for about 130 generations under either a constant environment (salt or cadmium), or an environment that is heterogeneous in time or space (combining salt and cadmium). They report a wealth of results, of which we summarize the most striking here. First, among genes that (i) were initially highly plastic and (ii) evolved significant divergence in expression levels between constant environment treatments, the evolved divergence is predominantly in the opposite direction to the initial plastic response. This suggests that either plasticity was initially maladaptive, or the selective pressure changed during the evolutionary process (see below). This somewhat unexpected result strikingly mirrors that from a study published last year in Nature [2], where the same pattern was found for responses of guppies to the presence of predators. However, Huang and Agrawal [1] went beyond this study by deciphering the underlying mechanisms in several interesting ways. First, they showed that change in gene expression often occurred at genes close to SNPs with differentiated frequencies across treatments (but not at genes with differentiated SNPs in their coding sequences), suggesting that cis-regulatory sequences are involved. This is also suggested by the fact that changes in gene expression are mostly caused by the increased expression of only one allele at polymorphic loci, and is a first step towards investigating the genetic underpinnings of (co)variation in gene expression levels. Another interesting set of findings concerns evolution of plasticity in treatments with variable environments. To compare the gene-expression plasticity that evolved in these treatments to an expectation, the authors considered that the expression levels in populations maintained for a long time under constant salt or cadmium had reached an optimum. The differences between these expression levels were thus assumed to predict the level of plasticity that should evolve in a heterogeneous environment (with both cadmium and salt) under perfect environmental predictability. The authors showed that plasticity did evolve more in the expected direction in heterogeneous than in constant environments, resulting in better adapted final expression levels across environments. Taken collectively, these results provide an unprecedented set of patterns that are greatly informative on how plasticity and evolution interact in constant versus changing environments. But of course, interpretations in terms of adaptive versus maladaptive plasticity are more challenging, as the authors themselves admit. Even though environmentally determined gene expression is the basic mechanism underlying the phenotypic plasticity of most traits, it is extremely difficult to relate to more integrated phenotypes for which we can understand the selection pressures, especially in multicellular organisms. The authors have recently investigated evolutionary change of quantitative traits in these selected lines, so it might be possible to establish links between reaction norms for macroscopic traits to those for gene expression levels. Such an approach would also involve tracking gene expression throughout life, rather than only in young larvae as done here, thus putting phenotypic complexity back in the picture also for expression levels. Another difficulty is that a plastic response that was originally adaptive may be replaced by an opposite evolutionary response in the long run, without having to invoke initially maladaptive plasticity. For instance, the authors mention the possibility that a generic stress response is initially triggered by cadmium, but is eventually unnecessary and costly after evolution of genetic mechanisms for cadmium detoxification (a case of so-called genetic accommodation). In any case, this study by Huang and Agrawal [1], together with the one by Ghalambor et al. last year [2], reports novel and unexpected results, which are likely to stimulate researchers interested in plasticity and evolution in heterogeneous environments for the years to come. References [1] Huang Y, Agrawal AF. 2016. Experimental Evolution of Gene Expression and Plasticity in Alternative Selective Regimes. PLoS Genetics 12:e1006336. doi: 10.1371/journal.pgen.1006336 [2] Ghalambor CK, Hoke KL, Ruell EW, Fischer EK, Reznick DN, Hughes KA. 2015. Non-adaptive plasticity potentiates rapid adaptive evolution of gene expression in nature. Nature 525: 372-375. doi: 10.1038/nature15256 | Experimental Evolution of Gene Expression and Plasticity in Alternative Selective Regimes | Huang Y, Agrawal AF | Little is known of how gene expression and its plasticity evolves as populations adapt to different environmental regimes. Expression is expected to evolve adaptively in all populations but only those populations experiencing environmental heterog... | | Adaptation, Experimental Evolution, Expression Studies, Phenotypic Plasticity | Luis-Miguel Chevin | 2016-12-20 09:04:15 | ||
04 Nov 2020
Treating symptomatic infections and the co-evolution of virulence and drug resistanceMore intense symptoms, more treatment, more drug-resistance: coevolution of virulence and drug-resistanceRecommended by Ludek Berec based on reviews by 3 anonymous reviewersMathematical models play an essential role in current evolutionary biology, and evolutionary epidemiology is not an exception [1]. While the issues of virulence evolution and drug-resistance evolution resonate in the literature for quite some time [2, 3], the study by Alizon [4] is one of a few that consider co-evolution of both these traits [5]. The idea behind this study is the following: treating individuals with more severe symptoms at a higher rate (which appears to be quite natural) leads to an appearance of virulent drug-resistant strains, via treatment failure. The author then shows that virulence in drug-resistant strains may face different selective pressures than in drug-sensitive strains and hence proceed at different rates. Hence, treatment itself modulates evolution of virulence. As one of the reviewers emphasizes, the present manuscript offers a mathematical view on why the resistant and more virulent strains can be selected in epidemics. Also, we both find important that the author highlights that the topic and results of this study can be attributed to public health policies and development of optimal treatment protocols [6]. References [1] Gandon S, Day T, Metcalf JE, Grenfell BT (2016) Forecasting epidemiological and evolutionary dynamics of infectious diseases. Trends Ecol Evol 31: 776-788. doi: https://doi.org/10.1016/j.tree.2016.07.010 | Treating symptomatic infections and the co-evolution of virulence and drug resistance | Samuel Alizon | <p>Antimicrobial therapeutic treatments are by definition applied after the onset of symptoms, which tend to correlate with infection severity. Using mathematical epidemiology models, I explore how this link affects the coevolutionary dynamics bet... | | Evolutionary Applications, Evolutionary Dynamics, Evolutionary Epidemiology, Evolutionary Theory | Ludek Berec | 2020-03-04 10:18:39 | ||
05 Jun 2018
The dynamics of preferential host switching: host phylogeny as a key predictor of parasite prevalence and distributionShift or stick? Untangling the signatures of biased host switching, and host-parasite co-speciationRecommended by Lucy Weinert based on reviews by Damien de Vienne and Nathan MeddMany emerging diseases arise by parasites switching to new host species, while other parasites seem to remain with same host lineage for very long periods of time, even over timescales where an ancestral host species splits into two or more new species. The ability to understand these dynamics would form an important part of our understanding of infectious disease. Experiments are clearly important for understanding these processes, but so are comparative studies, investigating the variation that we find in nature. Such comparative data do show strong signs of non-randomness, and this suggests that the epidemiological and ecological processes might be predictable, at least in part. For example, when we map patterns of parasite presence/absence onto host phylogenies, we often find that certain host clades harbour many more parasites than expected, or that closely-related hosts harbour closely-related parasites. Nevertheless, it remains difficult to interpret these patterns to make inferences about ecological and epidemiological processes. This is partly because non-random associations can arise in multiple ways. For example, parasites might be inherited from the common ancestor of related hosts, or might switch to new hosts, but preferentially establish on novel hosts that are closely related to their existing host. Infection might also influence the shape of host phylogeny, either by increasing the rate of host extinction or, conversely, increasing the rate of speciation (as with manipulative symbionts that might induce reproductive isolation). These various processes have, by and large, been studied in isolation, but the model introduced by Engelstädter and Fortuna [1], makes an important first step towards studying them together. Without such combined analyses, we will not be able to tell if the processes have their own unique signatures, or whether the same sort of non-randomness can arise in multiple ways. A major finding of the work is that the size of a host clade can be an important determinant of its overall infection level. This had been shown in previous work, assuming that the host phylogeny was fixed, but the current paper shows that it extends also to situations where host extinction and speciation takes place at a comparable rate to host shifting. This finding, then, calls into question the natural assumption that a clade of host species that is highly parasite ridden, must have some genetic or ecological characteristic that makes them particularly prone to infection, arguing that the clade size, rather than any characteristic of the clade members, might be the important factor. It will be interesting to see whether this prediction about clade size is borne out with comparative studies. Another feature of the study is that the framework is naturally extendable, to include further processes, such as the influence of parasite presence on extinction or speciation rates. No doubt extensions of this kind will form the basis of important future work. References [1] Engelstädter J and Fortuna NZ. 2018. The dynamics of preferential host switching: host phylogeny as a key predictor of parasite prevalence and distribution. bioRxiv 209254 ver. 5 peer-reviewed by Peer Community In Evolutionary Biology. doi: 10.1101/209254 | The dynamics of preferential host switching: host phylogeny as a key predictor of parasite prevalence and distribution | Jan Engelstaedter & Nicole Fortuna | <p>New parasites commonly arise through host-shifts, where parasites from one host species jump to and become established in a new host species. There is much evidence that the probability of host-shifts decreases with increasing phylogenetic dist... | | Bioinformatics & Computational Biology, Evolutionary Epidemiology, Evolutionary Theory, Macroevolution, Phylogenetics / Phylogenomics, Species interactions | Lucy Weinert | 2017-10-30 02:06:06 | ||
12 Feb 2024
How do plant RNA viruses overcome the negative effect of Muller s ratchet despite strong transmission bottlenecks?How to survive the mutational meltdown: lessons from plant RNA virusesRecommended by Kavita Jain based on reviews by Brent Allman, Ana Morales-Arce and 1 anonymous reviewerAlthough most mutations are deleterious, the strongly deleterious ones do not spread in a very large population as their chance of fixation is very small. Another mechanism via which the deleterious mutations can be eliminated is via recombination or sexual reproduction. However, in a finite asexual population, the subpopulation without any deleterious mutation will eventually acquire a deleterious mutation resulting in the reduction of the population size or in other words, an increase in the genetic drift. This, in turn, will lead the population to acquire deleterious mutations at a faster rate eventually leading to a mutational meltdown. This irreversible (or, at least over some long time scales) accumulation of deleterious mutations is especially relevant to RNA viruses due to their high mutation rate, and while the prior work has dealt with bacteriophages and RNA viruses, the study by Lafforgue et al. [1] makes an interesting contribution to the existing literature by focusing on plants. In this study, the authors enquire how despite the repeated increase in the strength of genetic drift, how the RNA viruses manage to survive in plants. Following a series of experiments and some numerical simulations, the authors find that as expected, after severe bottlenecks, the fitness of the population decreases significantly. But if the bottlenecks are followed by population expansion, the Muller’s ratchet can be halted due to the genetic diversity generated during population growth. They hypothesize this mechanism as a potential way by which the RNA viruses can survive the mutational meltdown. As a theoretician, I find this investigation quite interesting and would like to see more studies addressing, e.g., the minimum population growth rate required to counter the potential extinction for a given bottleneck size and deleterious mutation rate. Of course, it would be interesting to see in future work if the hypothesis in this article can be tested in natural populations. References [1] Guillaume Lafforgue, Marie Lefebvre, Thierry Michon, Santiago F. Elena (2024) How do plant RNA viruses overcome the negative effect of Muller s ratchet despite strong transmission bottlenecks? bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community In Evolutionary Biology | How do plant RNA viruses overcome the negative effect of Muller s ratchet despite strong transmission bottlenecks? | Guillaume Lafforgue, Marie Lefebvre, Thierry Michon, Santiago F. Elena | <p>Muller's ratchet refers to the irreversible accumulation of deleterious mutations in small populations, resulting in a decline in overall fitness. This phenomenon has been extensively observed in experiments involving microorganisms, including ... | | Experimental Evolution, Genome Evolution | Kavita Jain | 2023-08-04 09:37:08 | ||
26 Nov 2019
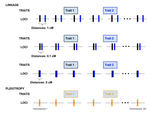
Pleiotropy or linkage? Their relative contributions to the genetic correlation of quantitative traits and detection by multi-trait GWA studiesUnderstanding the effects of linkage and pleiotropy on evolutionary adaptationRecommended by Kathleen Lotterhos based on reviews by Pär Ingvarsson and 1 anonymous reviewerGenetic correlations among traits are ubiquitous in nature. However, we still have a limited understanding of the genetic architecture of trait correlations. Some genetic correlations among traits arise because of pleiotropy - single mutations or genotypes that have effects on multiple traits. Other genetic correlations among traits arise because of linkage among mutations that have independent effects on different traits. Teasing apart the differential effects of pleiotropy and linkage on trait correlations is difficult, because they result in very similar genetic patterns. However, understanding these differential effects gives important insights into how ubiquitous pleiotropy may be in nature. References [1] Chebib, J. and Guillaume, F. (2019). Pleiotropy or linkage? Their relative contributions to the genetic correlation of quantitative traits and detection by multi-trait GWA studies. bioRxiv, 656413, v3 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/656413 | Pleiotropy or linkage? Their relative contributions to the genetic correlation of quantitative traits and detection by multi-trait GWA studies | Jobran Chebib and Frédéric Guillaume | <p>Genetic correlations between traits may cause correlated responses to selection depending on the source of those genetic dependencies. Previous models described the conditions under which genetic correlations were expected to be maintained. Sel... | | Bioinformatics & Computational Biology, Evolutionary Applications, Evolutionary Dynamics, Evolutionary Theory, Genome Evolution, Genotype-Phenotype, Molecular Evolution, Population Genetics / Genomics, Quantitative Genetics | Kathleen Lotterhos | 2019-06-05 13:51:43 | ||
11 Jun 2019

A bird’s white-eye view on neosex chromosome evolutionYoung sex chromosomes discovered in white-eye birdsRecommended by Kateryna Makova based on reviews by Gabriel Marais, Melissa Wilson and 1 anonymous reviewerRecent advances in next-generation sequencing are allowing us to uncover the evolution of sex chromosomes in non-model organisms. This study [1] represents an example of this application to birds of two Sylvioidea species from the genus Zosterops (commonly known as white-eyes). The study is exemplary in the amount and types of data generated and in the thoroughness of the analysis applied. Both male and female genomes were sequenced to allow the authors to identify sex-chromosome specific scaffolds. These data were augmented by generating the transcriptome (RNA-seq) data set. The findings after the analysis of these extensive data are intriguing: neoZ and neoW chromosome scaffolds and their breakpoints were identified. Novel sex chromosome formation appears to be accompanied by translocation events. The timing of formation of novel sex chromosomes was identified using molecular dating and appears to be relatively recent. Yet first signatures of distinct evolutionary patterns of sex chromosomes vs. autosomes could be already identified. These include the accumulation of transposable elements and changes in GC content. The changes in GC content could be explained by biased gene conversion and altered recombination landscape of the neo sex chromosomes. The authors also study divergence and diversity of genes located on the neo sex chromosomes. Here their findings appear to be surprising and need further exploration. The neoW chromosome already shows unique patterns of divergence and diversity at protein-coding genes as compared with genes on either neoZ or autosomes. In contrast, the genes on the neoZ chromosome do not display divergence or diversity patterns different from those for autosomes. This last observation is puzzling and I believe should be explored in further studies. Overall, this study significantly advances our knowledge of the early stages of sex chromosome evolution in vertebrates, provides an example of how such a study could be conducted in other non-model organisms, and provides several avenues for future work. References [1] Leroy T., Anselmetti A., Tilak M.K., Bérard S., Csukonyi L., Gabrielli M., Scornavacca C., Milá B., Thébaud C. and Nabholz B. (2019). A bird’s white-eye view on neo-sex chromosome evolution. bioRxiv, 505610, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/505610 | A bird’s white-eye view on neosex chromosome evolution | Thibault Leroy, Yoann Anselmetti, Marie-Ka Tilak, Sèverine Bérard, Laura Csukonyi, Maëva Gabrielli, Céline Scornavacca, Borja Milá, Christophe Thébaud, Benoit Nabholz | <p>Chromosomal organization is relatively stable among avian species, especially with regards to sex chromosomes. Members of the large Sylvioidea clade however have a pair of neo-sex chromosomes which is unique to this clade and originate from a p... | | Molecular Evolution, Population Genetics / Genomics | Kateryna Makova | 2019-01-24 14:17:15 | ||
27 Jul 2020
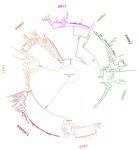
Evolution of the DAN gene family in vertebratesAn evolutionary view of a biomedically important gene familyRecommended by Kateryna Makova based on reviews by 2 anonymous reviewersThis manuscript [1] investigates the evolutionary history of the DAN gene family—a group of genes important for embryonic development of limbs, kidneys, and left-right axis speciation. This gene family has also been implicated in a number of diseases, including cancer and nephropathies. DAN genes have been associated with the inhibition of the bone morphogenetic protein (BMP) signaling pathway. Despite this detailed biochemical and functional knowledge and clear importance for development and disease, evolution of this gene family has remained understudied. The diversification of this gene family was investigated in all major groups of vertebrates. The monophyly of the gene members belonging to this gene family was confirmed. A total of five clades were delineated, and two novel lineages were discovered. The first lineage was only retained in cephalochordates (amphioxus), whereas the second one (GREM3) was retained by cartilaginous fish, holostean fish, and coelanth. Moreover, the patterns of chromosomal synteny in the chromosomal regions harboring DAN genes were investigated. Additionally, the authors reconstructed the ancestral gene repertoires and studied the differential retention/loss of individual gene members across the phylogeny. They concluded that the ancestor of gnathostome vertebrates possessed eight DAN genes that underwent differential retention during the evolutionary history of this group. During radiation of vertebrates, GREM1, GREM2, SOST, SOSTDC1, and NBL1 were retained in all major vertebrate groups. At the same time, GREM3, CER1, and DAND5 were differentially lost in some vertebrate lineages. At least two DAN genes were present in the common ancestor of vertebrates, and at least three DAN genes were present in the common ancestor of chordates. Therefore the patterns of retention and diversification in this gene family appear to be complex. Evolutionary slowdown for the DAN gene family was observed in mammals, suggesting selective constraints. Overall, this article puts the biomedical importance of the DAN family in the evolutionary perspective. References [1] Opazo JC, Hoffmann FG, Zavala K, Edwards SV (2020) Evolution of the DAN gene family in vertebrates. bioRxiv, 794404, ver. 3 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/794404 | Evolution of the DAN gene family in vertebrates | Juan C. Opazo, Federico G. Hoffmann, Kattina Zavala, Scott V. Edwards | <p>The DAN gene family (DAN, Differential screening-selected gene Aberrant in Neuroblastoma) is a group of genes that is expressed during development and plays fundamental roles in limb bud formation and digitation, kidney formation and morphogene... | | Molecular Evolution | Kateryna Makova | 2019-10-15 16:43:13 | ||
04 Mar 2024
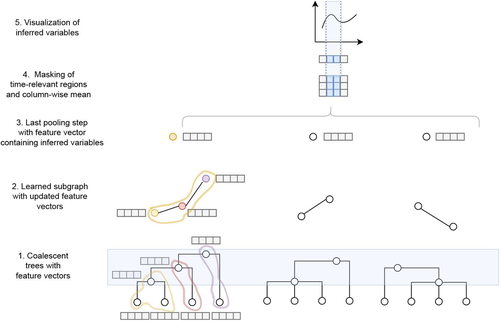
Simultaneous Inference of Past Demography and Selection from the Ancestral Recombination Graph under the Beta CoalescentBeyond the standard coalescent: demographic inference with complete genomes and graph neural networks under the beta coalescentRecommended by Julien Yann Dutheil based on reviews by 2 anonymous reviewers based on reviews by 2 anonymous reviewers
Modelling the evolution of complete genome sequences in populations requires accounting for the recombination process, as a single tree can no longer describe the underlying genealogy. The sequentially Markov coalescent (SMC, McVean and Cardin 2005; Marjoram and Wall 2006) approximates the standard coalescent with recombination process and permits estimating population genetic parameters (e.g., population sizes, recombination rates) using population genomic datasets. As such datasets become available for an increasing number of species, more fine-tuned models are needed to encompass the diversity of life cycles of organisms beyond the model species on which most methods have been benchmarked. The work by Korfmann et al. (Korfmann et al. 2024) represents a significant step forward as it accounts for multiple mergers in SMC models. Multiple merger models account for simultaneous coalescence events so that more than two lineages find a common ancestor in a given generation. This feature is not allowed in standard coalescent models and may result from selection or skewed offspring distributions, conditions likely met by a broad range of species, particularly microbial. Yet, this work goes beyond extending the SMC, as it introduces several methodological innovations. The "classical" SMC-based inference approaches rely on hidden Markov models to compute the likelihood of the data while efficiently integrating over the possible ancestral recombination graphs (ARG). Following other recent works (e.g. Gattepaille et al. 2016), Korfmann et al. propose to separate the ARG inference from model parameter estimation under maximum likelihood (ML). They introduce a procedure where the ARG is first reconstructed from the data and then taken as input in the model fitting step. While this approach does not permit accounting for the uncertainty in the ARG reconstruction (which is typically large), it potentially allows for the extraction of more information from the ARG, such as the occurrence of multiple merging events. Going away from maximum likelihood inference, the authors trained a graph neural network (GNN) on simulated ARGs, introducing a new, flexible way to estimate population genomic parameters. The authors used simulations under a beta-coalescent model with diverse demographic scenarios and showed that the ML and GNN approaches introduced can reliably recover the simulated parameter values. They further show that when the true ARG is given as input, the GNN outperforms the ML approach, demonstrating its promising power as ARG reconstruction methods improve. In particular, they showed that trained GNNs can disentangle the effects of selective sweeps and skewed offspring distributions while inferring past population size changes. This work paves the way for new, exciting applications, though many questions must be answered. How frequent are multiple mergers? As the authors showed that these events "erase" the record of past demographic events, how many genomes are needed to conduct reliable inference, and can the methods computationally cope with the resulting (potentially large) amounts of required data? This is particularly intriguing as micro-organisms, prone to strong selection and skewed offspring distributions, also tend to carry smaller genomes. References Gattepaille L, Günther T, Jakobsson M. 2016. Inferring Past Effective Population Size from Distributions of Coalescent Times. Genetics 204:1191-1206. | Simultaneous Inference of Past Demography and Selection from the Ancestral Recombination Graph under the Beta Coalescent | Kevin Korfmann, Thibaut Sellinger, Fabian Freund, Matteo Fumagalli, Aurélien Tellier | <p style="text-align: justify;">The reproductive mechanism of a species is a key driver of genome evolution. The standard Wright-Fisher model for the reproduction of individuals in a population assumes that each individual produces a number of off... | | Adaptation, Bioinformatics & Computational Biology, Evolutionary Applications, Evolutionary Theory, Life History, Population Genetics / Genomics | Julien Yann Dutheil | 2023-07-31 13:11:22 |




