
Latest recommendations

| Id | Title▼ | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
24 Mar 2023
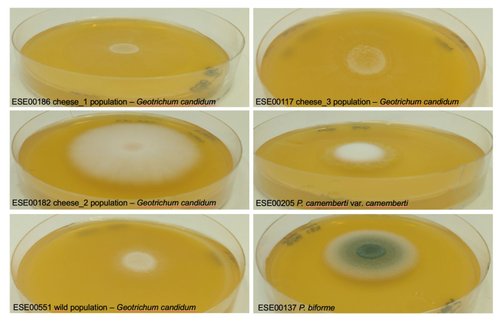
Domestication of different varieties in the cheese-making fungus Geotrichum candidumDiverse outcomes in cheese fungi domesticationRecommended by Christelle Fraïsse based on reviews by Delphine Sicard and 1 anonymous reviewer based on reviews by Delphine Sicard and 1 anonymous reviewer
Domestication is a complex process that imprints the demography and the genomes of domesticated populations, enforcing strong selective pressures on traits favourable to humans, e.g. for food production [1]. Domestication has been quite intensely studied in plants and animals, but less so in micro-organisms such as fungi, despite their assets (e.g. their small genomes and tractability in the lab). This elegant study by Bennetot and collaborators [2] on the cheese-making fungus Geotrichum candidum adds to the mounting body of studies in the genomics of fungi, proving they are excellent models in evolutionary biology for studying adaptation and drift in eukaryotes [3]. Bennetot et al. newly showed with whole genome sequences that all G. candidum strains isolated from cheese form a monophyletic clade subdivided into three genetically differentiated populations with several admixed strains, while the wild strains sampled from diverse geographic locations form a sister clade. This suggests the wild progenitor was not sampled in the present study and calls for future exciting work on the domestication history of the G. candidum fungus. The authors scanned the genomes for footprints of adaptation to the cheese environment and identified promising candidates, such as a gene involved in iron uptake (this element is limiting in cheese). Their functional genome analysis also provides evidence for higher contents of transposable elements in cheese-making strains, likely due to relaxed selection during the domestication process. This paper is particularly impressive in that the authors complemented the population genomic approach with the phenotypic characterization of the strains and tested their ability to outcompete common fungal food spoilers. The authors convincingly showed that cheese-making strains display phenotypic differences relative to wild relatives for multiple traits such as slower growth, lower proteolysis activity and a greater amount of volatiles attractive to consumers, these phenotypes being beneficial for cheese making. Finally, this work is particularly inspiring because it thoroughly discusses convergent evolution during domestication in different cheese-associated fungi. Indeed, studying populations experiencing similar environmental pressures is fundamental to understanding whether evolution is repeatable [4]. For instance, all three cheese populations of G. candidum exhibit a lower genetic diversity than wild populations. However, only one population displays a stronger domestication syndrome, resembling the Penicillium camemberti situation [5]. Furthermore, different cheese-making practices may have led to varying situations with clonal lineages in non-Roquefort P. roqueforti and P. camemberti [5, 6], while the cheese-making G. candidum populations still harbour some diversity. In a nutshell, Bennetot's study makes an important contribution to evolutionary biology and highlights the value of diversifying our model organisms toward under-represented clades. REFERENCES [1] Diamond J (2002) Evolution, consequences and future of plant and animal domestication. Nature 418: 700–707. https://doi.org/10.1038/nature01019 [2] Bennetot B, Vernadet J-P, Perkins V, Hautefeuille S, Rodríguez de la Vega RC, O’Donnell S, Snirc A, Grondin C, Lessard M-H, Peron A-C, Labrie S, Landaud S, Giraud T, Ropars J (2023) Domestication of different varieties in the cheese-making fungus Geotrichum candidum. bioRxiv, 2022.05.17.492043, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.05.17.492043 [3] Gladieux P, Ropars J, Badouin H, Branca A, Aguileta G, de Vienne DM, Rodríguez de la Vega RC, Branco S, Giraud T (2014) Fungal evolutionary genomics provides insight into the mechanisms of adaptive divergence in eukaryotes. Mol. Ecol. 23: 753–773. https://doi.org/10.1111/mec.12631 [4] Bolnick DI, Barrett RD, Oke KB, Rennison DJ, Stuart YE (2018) (Non)Parallel evolution. Ann. Rev. Ecol. Evol. Syst. 49: 303–330. https://doi.org/10.1146/annurev-ecolsys-110617-062240 [5] Ropars J, Didiot E, Rodríguez de la Vega RC, Bennetot B, Coton M, Poirier E, Coton E, Snirc A, Le Prieur S, Giraud T (2020) Domestication of the Emblematic White Cheese-Making Fungus Penicillium camemberti and Its Diversification into Two Varieties. Current Biol. 30: 4441–4453.e4. https://doi.org/10.1016/j.cub.2020.08.082 [6] Dumas, E, Feurtey, A, Rodríguez de la Vega, RC, Le Prieur S, Snirc A, Coton M, Thierry A, Coton E, Le Piver M, Roueyre D, Ropars J, Branca A, Giraud T (2020) Independent domestication events in the blue-cheese fungus Penicillium roqueforti. Mol Ecol. 29: 2639–2660. https://doi.org/10.1111/mec.15359 | Domestication of different varieties in the cheese-making fungus *Geotrichum candidum* | Bastien Bennetot, Jean-Philippe Vernadet, Vincent Perkins, Sophie Hautefeuille, Ricardo C. Rodríguez de la Vega, Samuel O’Donnell, Alodie Snirc, Cécile Grondin, Marie-Hélène Lessard, Anne-Claire Peron, Steve Labrie, Sophie Landaud, Tatiana Giraud,... | <p>Domestication is an excellent model for studying adaptation processes, involving recent adaptation and diversification, convergence following adaptation to similar conditions, as well as degeneration of unused functions. <em>Geotrichum candidum... | | Adaptation, Genome Evolution, Population Genetics / Genomics | Christelle Fraïsse | 2022-08-12 20:50:42 | ||
05 Apr 2024
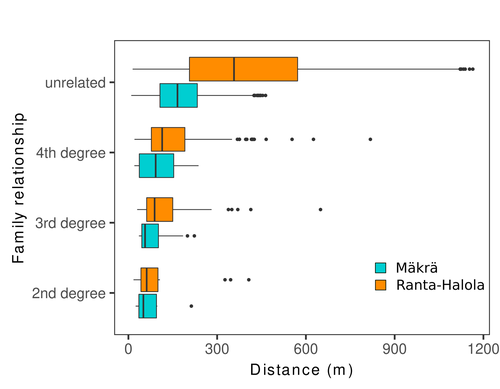
Does the seed fall far from the tree? Weak fine scale genetic structure in a continuous Scots pine populationWeak spatial genetic structure in a large continuous Scots pine population – implications for conservation and breedingRecommended by Myriam Heuertz based on reviews by Joachim Mergeay, Jean-Baptiste Ledoux and Roberta Loh
Spatial genetic structure, i.e. the non-random spatial distribution of genotypes, arises in populations because of different processes including spatially limited dispersal and selection. Knowledge on the spatial genetic structure of plant populations is important to assess biological parameters such as gene dispersal distances and the potential for local adaptations, as well as for applications in conservation management and breeding. In their work, Niskanen and colleagues demonstrate a multifaceted approach to characterise the spatial genetic structure in two replicate sites of a continuously distributed Scots pine population in South-Eastern Finland. They mapped and assessed the ages of 469 naturally regenerated adults and genotyped them using a SNP array which resulted in 157 325 filtered polymorphic SNPs. Their dataset is remarkably powerful because of the large numbers of both individuals and SNPs genotyped. This made it possible to characterise precisely the decay of genetic relatedness between individuals with spatial distance despite the extensive dispersal capacity of Scots pine through pollen, and ensuing expectations of an almost panmictic population. The authors’ data analysis was particularly thorough. They demonstrated that two metrics of pairwise relatedness, the genomic relationship matrix (GRM, Yang et al. 2011) and the kinship coefficient (Loiselle et al. 1995) were strongly correlated and produced very similar inference of family relationships: >99% of pairs of individuals were unrelated, and the remainder exhibited 2nd (e.g., half-siblings) to 4th degree relatedness. Pairwise relatedness decayed with spatial distance which resulted in extremely weak but statistically significant spatial genetic structure in both sites, quantified as Sp=0.0005 and Sp=0.0008. These estimates are at least an order of magnitude lower than estimates in the literature obtained in more fragmented populations of the same species or in other conifers. Estimates of the neighbourhood size, the effective number of potentially mating individuals belonging to a within-population neighbourhood (Wright 1946), were relatively large with Nb=1680-3210 despite relatively short gene dispersal distances, σg = 36.5–71.3m, which illustrates the high effective density of the population. The authors showed the implications of their findings for selection. The capacity for local adaptation depends on dispersal distances and the strength of the selection coefficient. In the study population, the authors inferred that local adaptation can only occur if environmental heterogeneity occurs over a distance larger than approximately one kilometre (or larger, if considering long-distance dispersal). Interestingly, in Scots pine, no local adaptation has been described on similar geographic scales, in contrast to some other European or Mediterranean conifers (Scotti et al. 2023). The authors’ results are relevant for the management of conservation and breeding. They showed that related individuals occurred within sites only and that they shared a higher number of rare alleles than unrelated ones. Since rare alleles are enriched in new and recessive deleterious variants, selecting related individuals could have negative consequences in breeding programmes. The authors also showed, in their response to reviewers, that their powerful dataset was not suitable to obtain a robust estimate of effective population size, Ne, based on the linkage disequilibrium method (Do et al. 2014). This illustrated that the estimation of Ne used for genetic indicators supported in international conservation policy (Hoban et al. 2020, CBD 2022) remains challenging in large and continuous populations (see also Santo-del-Blanco et al. 2023, Gargiulo et al. 2024). ReferencesCBD (2022) Kunming-Montreal Global Biodiversity Framework. https://www.cbd.int/doc/decisions/cop-15/cop-15-dec-04-en.pdf Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014). NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne ) from genetic data. Molecular Ecology Resources 14: 209–214. https://doi.org/10.1111/1755-0998.12157 Gargiulo R, Decroocq V, González-Martínez SC, Paz-Vinas I, Aury JM, Kupin IL, Plomion C, Schmitt S, Scotti I, Heuertz M (2024) Estimation of contemporary effective population size in plant populations: limitations of genomic datasets. Evolutionary Applications, in press, https://doi.org/10.1101/2023.07.18.549323 Hoban S, Bruford M, D’Urban Jackson J, Lopes-Fernandes M, Heuertz M, Hohenlohe PA, Paz-Vinas I, et al. (2020) Genetic diversity targets and indicators in the CBD post-2020 Global Biodiversity Framework must be improved. Biological Conservation 248: 108654. https://doi.org/10.1016/j.biocon.2020.108654 Loiselle BA, Sork VL, Nason J & Graham C (1995) Spatial genetic structure of a tropical understorey shrub, Psychotria officinalis (Rubiaceae). American Journal of Botany 82: 1420–1425. https://doi.org/10.1002/j.1537-2197.1995.tb12679.x Santos-del-Blanco L, Olsson S, Budde KB, Grivet D, González-Martínez SC, Alía R, Robledo-Arnuncio JJ (2022). On the feasibility of estimating contemporary effective population size (Ne) for genetic conservation and monitoring of forest trees. Biological Conservation 273: 109704. https://doi.org/10.1016/j.biocon.2022.109704 Scotti I, Lalagüe H, Oddou-Muratorio S, Scotti-Saintagne C, Ruiz Daniels R, Grivet D, et al. (2023) Common microgeographical selection patterns revealed in four European conifers. Molecular Ecology 32: 393-411. https://doi.org/10.1111/mec.16750 Wright S (1946) Isolation by distance under diverse systems of mating. Genetics 31: 39–59. https://doi.org/10.1093/genetics/31.1.39 Yang J, Lee SH, Goddard ME & Visscher PM (2011) GCTA: a tool for genome-wide complex trait analysis. The American Journal of Human Genetics 88: 76–82. https://www.cell.com/ajhg/pdf/S0002-9297(10)00598-7.pdf | Does the seed fall far from the tree? Weak fine scale genetic structure in a continuous Scots pine population | Alina K. Niskanen, Sonja T. Kujala, Katri Kärkkäinen, Outi Savolainen, Tanja Pyhäjärvi | <p>Knowledge of fine-scale spatial genetic structure, i.e., the distribution of genetic diversity at short distances, is important in evolutionary research and in practical applications such as conservation and breeding programs. In trees, related... | | Adaptation, Evolutionary Applications, Population Genetics / Genomics | Myriam Heuertz | Joachim Mergeay | 2023-06-27 21:57:28 | |
29 Nov 2023

Does sociality affect evolutionary speed?On the evolutionary implications of being a social animalRecommended by Michael D Greenfield based on reviews by Rafael Lucas Rodriguez and 1 anonymous reviewerWhat does it mean to be highly social? Considering the so-called four ‘pinnacles’ of animal society (Wilson, 1975) – humans, cooperative breeding as found in some non-human mammals and birds, the social insects, and colonial marine invertebrates – having inter-individual relations extending beyond the sexual pair and the parent-offspring interaction is foremost. In many cases being social implies a high local population density, interaction with the same group of individuals over an extended time period, and an overlapping of generations. Additional features of social species may be a wide geographical range, perhaps associated with ecological and behavioral plasticity, the latter often facilitated by cultural transmission of traditions. Narrowing our perspective to the domain of PCI Evolutionary Biology, we might continue our question by asking whether being social predisposes one to a special evolutionary path toward the future. Do social species evolve faster (or slower) than their more solitary relatives such that over time they are more unlike (or similar to) those relatives (anagenesis)? And are evolutionary changes in social species more or less likely to be accompanied by lineage splitting (cladogenesis) and ultimately speciation? The latter question is parallel to one first posed over 40 years ago (West-Eberhard, 1979; Lande, 1981) for sexually selected traits: Do strong mating preferences and conspicuous courtship signals generate speciation via the Fisherian process or ecological divergence? An extensive survey of birds had found little supporting evidence (Price, 1998), but a recent one that focused on plumage complexity in tanagers did reveal a relationship, albeit a weak one (Price-Waldman et al., 2020). Because sexual selection has been viewed as a part of the broader process of social selection (West-Eberhard, 1979), it is thus fitting to extend our surveys to the evolutionary implications of being social. Unlike the inquiry for a sexual selection - evolutionary change connection, a social behavior counterpart has remained relatively untreated. Diverse logistical problems might account for this oversight. What objective proxies can be used for social behavior, and for the rate of evolutionary change within a lineage? How many empirical studies have generated data from which appropriate proxies could be extracted? More intractable is the conundrum arising from the connectedness between socially- and sexually-selected traits. For example, the elevated population density found in highly social species can greatly increase the mating advantage enjoyed by an attractive male. If anagenesis is detected, did it result from social behavior or sexual selection? And if social behavior leads to a group structure in which male-male competition is reduced, would a modest rate of evolutionary change be support for the sexual selection - evolutionary speed connection or evidence opposing the sociality - evolution one? Against the above odds, several biologists have begun to explore the notion that social behavior just might favor evolutionary speed in either anagenesis or cladogenesis. In a recent analysis relying on the comparative method, Lluís Socias-Martínez and Louise Rachel Peckre (2023) combed the scientific literature archives and identified those studies with specific data on the relationships between sexual selection or social behavior and evolutionary change, either anagenesis or cladogenesis. The authors were careful to employ fairly conservative criteria for including studies, and the number eventually retained was small. Nonetheless, some patterns emerge: Many more studies report anagenesis than cladogenesis, and many more report correlations with sexually-selected traits than with non-sexual social behavior ones. And, no study indicates a potential effect of social behavior on cladogenesis. Is this latter observation authentic or an artifact of a paucity of data? There are some a priori reasons why cladogenesis may seldom arise. Whereas highly social behavior could lead to fission encompassing mutually isolated population clusters within a species, social behavior may also engender counterbalancing plasticity that allows and even promotes inter-cluster migration and fusion. And briefly – and non-systematically, as the rate of lineage splitting would need to be measured – looking at one of the pinnacles of animal social behavior, the social insects, there is little indication that diversification has been accelerated. There are fewer than 3000 described species of termites, only ca. 16,000 ants, and the vast majority of bees and wasps are solitary. Lluís Socias-Martínez and Louise Rachel Peckre provide us with a very detailed discussion of these and a myriad of other complications. I end with a common refrain, we need more consideration of the authors’ interesting question, and much more data and analysis. One can thank Socias-Martínez and Peckre for pointing us in that direction. References Lande, R. (1981). Models of speciation by sexual selection on polygenic traits. Proc. Natn. Acad. Sci. USA 78, 3721-3725. https://doi.org/10.1073/pnas.78.6.3721 Price, T. (1998). Sexual selection and natural selection in bird speciation. Phil. Trans. Roy. Soc. B, 353, 251-260. https://doi.org/10.1098/rstb.1998.0207 Price‐Waldman, R. M., Shultz, A. J., & Burns, K. J. (2020). Speciation rates are correlated with changes in plumage color complexity in the largest family of songbirds. Evolution, 74(6), 1155–1169. https://doi.org/10.1111/evo.13982 Socias-Martínez and Peckre. (2023). Does sociality affect evolutionary speed? Zenodo, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.5281/zenodo.10086186 West-Eberhard, M. J. (1979). Sexual selection, social competition, and evolution. Proceedings of the American Philosophical Society, 123(4), 222–234. http://www.jstor.org/stable/2828804 Wilson, E. O. (1975). Sociobiology. The New Synthesis. Cambridge, Mass., The Belknap Press of Harvard University | Does sociality affect evolutionary speed? | Lluís Socias-Martínez, Louise Rachel Peckre | <p>An overlooked source of variation in evolvability resides in the social lives of animals. In trying to foster research in this direction, we offer a critical review of previous work on the link between evolutionary speed and sociality. A first ... | | Behavior & Social Evolution, Evolutionary Dynamics, Evolutionary Theory, Genome Evolution, Macroevolution, Molecular Evolution, Population Genetics / Genomics, Sexual Selection, Speciation | Michael D Greenfield | 2023-03-03 00:10:49 | ||
05 Oct 2022
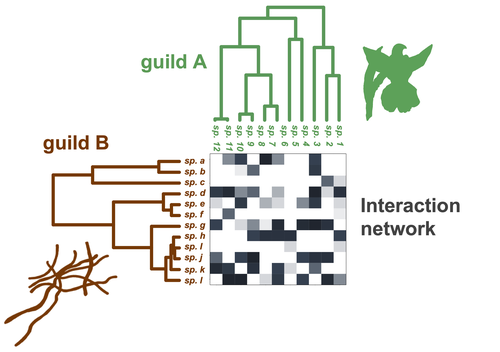
Do closely related species interact with similar partners? Testing for phylogenetic signal in bipartite interaction networksTesting for phylogenetic signal in species interaction networksRecommended by Alejandro Gonzalez Voyer based on reviews by Joaquin Calatayud and Thomas GuillermeSpecies are immersed within communities in which they interact mutualistically, as in pollination or seed dispersal, or nonreciprocally, such as in predation or parasitism, with other species and these interactions play a paramount role in shaping biodiversity (Bascompte and Jordano 2013). Researchers have become increasingly interested in the processes that shape these interactions and how these influence community structure and responses to disturbances. Species interactions are often described using bipartite interaction networks and one important question is how the evolutionary history of the species involved influences the network, including whether there is phylogenetic signal in interactions, in other words whether closely related species interact with other closely related species (Bascompte and Jordano 2013, Perez-Lamarque et al. 2022). To address this question different approaches, correlative and model-based, have been developed to test for phylogenetic signal in interactions, although comparative analyses of the performance of these different metrics are lacking. In their article Perez-Lamarque et al. (2022) set out to test the statistical performance of two widely-used methods, Mantel tests and Phylogenetic Bipartite Linear Models (PBLM; Ives and Godfray 2006) using simulations. Phylogenetic signal is measured as the degree to which distance to the nearest common ancestor predicts the observed similarity in trait values among species. In species interaction networks, the data are actually the between-species dissimilarity among interacting species (Perez-Lamarque et al. 2022), and typical approaches to test for phylogenetic signal cannot be used. However, the Mantel test provides a useful means of analyzing the correlation between two distance matrices, the between-species phylogenetic distance and the between-species dissimilarity in interactions. The PBLM approach, on the other hand, assumes that interactions between species are influenced by unobserved traits that evolve along the phylogenies following a given phenotypic evolution model and the parameters of this model are interpreted in terms of phylogenetic signal (Ives and Godfray 2006). Perez-Lamarque et al (2022) found that the model-based PBLM approach has a high type-I error rate, in other words it often detected phylogenetic signal when there was none. The simple Mantel test was found to present a low type-I error rate and moderate statistical power. However, it tended to overestimate the degree to which species interact with dissimilar partners. In addition to the aforementioned analyses, the authors also tested whether the simple Mantel test was able to detect phylogenetic signal in interactions among species within a given clade in the phylogeny, as phylogenetic signal in species interactions may be localized within specific clades. The article concludes with general guidelines for users wishing to test phylogenetic signal in their interaction networks and illustrates them with an example of an orchid-mycorrhizal fungus network from the oceanic island of La Réunion (Martos et al 2012). This broadly accessible article provides a valuable analysis of the performance of tests of phylogenetic signal in interaction networks enabling users to make informed choices of the analytical methods they wish to employ, and provide useful and detailed guidelines. Therefore, the work should be of broad interest to researchers studying species interactions. References Bascompte J, Jordano P (2013) Mutualistic Networks. Princeton University Press. https://doi.org/10.1515/9781400848720 Ives AR, Godfray HCJ (2006) Phylogenetic Analysis of Trophic Associations. The American Naturalist, 168, E1–E14. https://doi.org/10.1086/505157 Martos F, Munoz F, Pailler T, Kottke I, Gonneau C, Selosse M-A (2012) The role of epiphytism in architecture and evolutionary constraint within mycorrhizal networks of tropical orchids. Molecular Ecology, 21, 5098–5109. https://doi.org/10.1111/j.1365-294X.2012.05692.x Perez-Lamarque B, Maliet O, Pichon B, Selosse M-A, Martos F, Morlon H (2022) Do closely related species interact with similar partners? Testing for phylogenetic signal in bipartite interaction networks. bioRxiv, 2021.08.30.458192, ver. 6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.08.30.458192 | Do closely related species interact with similar partners? Testing for phylogenetic signal in bipartite interaction networks | Benoît Perez-Lamarque, Odile Maliet, Benoît Pichon, Marc-André Selosse, Florent Martos, Hélène Morlon | <p style="text-align: justify;">Whether interactions between species are conserved on evolutionary time-scales has spurred the development of both correlative and process-based approaches for testing phylogenetic signal in interspecific interactio... | | Evolutionary Ecology, Species interactions | Alejandro Gonzalez Voyer | 2022-03-10 13:48:15 | ||
16 Nov 2022

Divergence of olfactory receptors associated with the evolution of assortative mating and reproductive isolation in miceTinder in mice: A match made with the sense of smellRecommended by Christelle Fraïsse based on reviews by Angeles de Cara, Ludovic Claude Maisonneuve and 1 anonymous reviewer
Differentiation-based genome scans lie at the core of speciation and adaptation genomics research. Dating back to Lewontin & Krakauer (1973), they have become very popular with the advent of genomics to identify genome regions of enhanced differentiation relative to neutral expectations. These regions may represent genetic barriers between divergent lineages and are key for studying reproductive isolation. However, genome scan methods can generate a high rate of false positives, primarily if the neutral population structure is not accounted for (Bierne et al. 2013). Moreover, interpreting genome scans can be challenging in the context of secondary contacts between diverging lineages (Bierne et al. 2011), because the coupling between different components of reproductive isolation (local adaptation, intrinsic incompatibilities, mating preferences, etc.) can occur readily, thus preventing the causes of differentiation from being determined. Smadja and collaborators (2022) applied a sophisticated genome scan for trait association (BAYPASS, Gautier 2015) to underlie the genetic basis of a polygenetic behaviour: assortative mating in hybridizing mice. My interest in this neat study mainly relies on two reasons. First, the authors used an ingenious geographical setting (replicate pairs of “Choosy” versus “Non-Choosy” populations) with multi-way comparisons to narrow down the list of candidate regions resulting from BAYPASS. The latter corrects for population structure, handles cost-effective pool-seq data and allows for gene-based analyses that aggregate SNP signals within a gene. These features reinforce the set of outlier genes detected; however, not all are expected to be associated with mating preference. The second reason why this study is valuable to me is that Smadja et al. (2022) complemented the population genomic approach with functional predictions to validate the genetic signal. In line with previous behavioural and chemical assays on the proximal mechanisms of mating preferences, they identified multiple olfactory and vomeronasal receptor genes as highly significant candidates. Therefore, combining genomic signals with functional analyses is a clever way to provide insights into the causes of reproductive isolation, especially when multiple barriers are involved. This is typically true for reinforcement (Butlin & Smadja 2018), suspected to occur in these mice because, in that case, assortative mating (a prezygotic barrier) evolves in response to the cost of hybridization (for example, due to hybrid inviability). As advocated by the authors, their study paves the way for future work addressing the genetic basis of reinforcement, a trait of major evolutionary importance for which we lack empirical data. They also make a compelling case using complementary approaches that olfactory and vomeronasal receptors have a central role in mammal speciation.
Bierne N, Welch J, Loire E, Bonhomme F, David P (2011) The coupling hypothesis: why genome scans may fail to map local adaptation genes. Mol Ecol 20: 2044–2072. https://doi.org/10.1111/j.1365-294X.2011.05080.x Bierne N, Roze D, Welch JJ (2013) Pervasive selection or is it…? why are FST outliers sometimes so frequent? Mol Ecol 22: 2061–2064. https://doi.org/10.1111/mec.12241 Butlin RK, Smadja CM (2018) Coupling, Reinforcement, and Speciation. Am Nat 191:155–172. https://doi.org/10.1086/695136 Gautier M (2015) Genome-Wide Scan for Adaptive Divergence and Association with Population-Specific Covariates. Genetics 201:1555–1579. https://doi.org/10.1534/genetics.115.181453 Lewontin RC, Krakauer J (1973) Distribution of gene frequency as a test of the theory of selective neutrality of polymorphisms. Genetics 74: 175–195. https://doi.org/10.1093/genetics/74.1.175 Smadja CM, Loire E, Caminade P, Severac D, Gautier M, Ganem G (2022) Divergence of olfactory receptors associated with the evolution of assortative mating and reproductive isolation in mice. bioRxiv, 2022.07.21.500634, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.07.21.500634 | Divergence of olfactory receptors associated with the evolution of assortative mating and reproductive isolation in mice | Carole M. Smadja, Etienne Loire, Pierre Caminade, Dany Severac, Mathieu Gautier, Guila Ganem | <p>Deciphering the genetic bases of behavioural traits is essential to understanding how they evolve and contribute to adaptation and biological diversification, but it remains a substantial challenge, especially for behavioural traits with polyge... | | Adaptation, Behavior & Social Evolution, Genotype-Phenotype, Speciation | Christelle Fraïsse | 2022-07-25 11:54:52 | ||
20 Nov 2019
Distribution of iridescent colours in hummingbird communities results from the interplay between selection for camouflage and communicationFeathers iridescence sheds light on the assembly rules of humingbirds communitiesRecommended by Sébastien Lavergne based on reviews by 2 anonymous reviewersEcology needs rules stipulating how species distributions and ecological communities should be assembled along environmental gradients, but few rules have yet emerged in the ecological literature. The search of ecogeographical rules governing the spatial variation of birds colours has recently known an upsurge of interest in the litterature [1]. Most studies have, however, looked at pigmentary colours and not structural colours (e.g. iridescence), although it is know that color perception by animals (both birds and their predators) can be strongly influenced by light diffraction causing iridescence patterns on feathers. References [1] Delhey, K. (2019). A review of Gloger’s rule, an ecogeographical rule of colour: definitions, interpretations and evidence. Biological Reviews, 94(4), 1294–1316. doi: 10.1111/brv.12503 | Distribution of iridescent colours in hummingbird communities results from the interplay between selection for camouflage and communication | Hugo Gruson, Marianne Elias, Juan L. Parra, Christine Andraud, Serge Berthier, Claire Doutrelant, Doris Gomez | <p>Identification errors between closely related, co-occurring, species may lead to misdirected social interactions such as costly interbreeding or misdirected aggression. This selects for divergence in traits involved in species identification am... | | Evolutionary Ecology, Macroevolution, Phylogeography & Biogeography, Sexual Selection, Species interactions | Sébastien Lavergne | 2019-03-29 17:23:20 | ||
14 Feb 2024
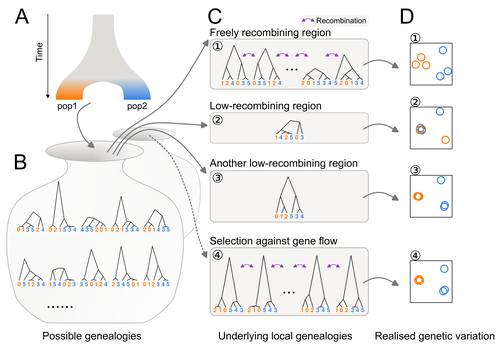
Distinct patterns of genetic variation at low-recombining genomic regions represent haplotype structureDiscerning the causes of local deviations in genetic variation: the effect of low-recombination regionsRecommended by Matteo Fumagalli based on reviews by Claire Merot and 1 anonymous reviewer
In this study, Ishigohoka and colleagues tackle an important, yet often overlooked, question on the causes of genetic variation. While genome-wide patterns represent population structure, local variation is often associated with selection. Authors propose that an alternative cause for variation in individual loci is reduced recombination rate. To test this hypothesis, authors perform local Principal Component Analysis (PCA) (Li & Ralph, 2019) to identify local deviations in population structure in the Eurasian blackcap (Sylvia atricapilla) (Ishigohoka et al. 2022). This approach is typically used to detect chromosomal rearrangements or any long region of linked loci (e.g., due to reduced recombination or selection) (Mérot et al. 2021). While other studies investigated the effect of low recombination on genetic variation (Booker et al. 2020), here authors provide a comprehensive analysis of the effect of recombination to local PCA patterns both in empirical and simulated data sets. Findings demonstrate that low recombination (and not selection) can be the sole explanatory variable for outlier windows. The study also describes patterns of genetic variation along the genome of Eurasian blackcaps, localising at least two polymorphic inversions (Ishigohoka et al. 2022). Further investigations on the effect of model parameters (e.g., window sizes and thresholds for defining low-recombining regions), as well as the use of powerful neutrality tests are in need to clearly assess whether outlier regions experience selection and reduced recombination, and to what extent. References Booker, T. R., Yeaman, S., & Whitlock, M. C. (2020). Variation in recombination rate affects detection of outliers in genome scans under neutrality. Molecular Ecology, 29 (22), 4274–4279. https://doi.org/10.1111/mec.15501 Ishigohoka, J., Bascón-Cardozo, K., Bours, A., Fuß, J., Rhie, A., Mountcastle, J., Haase, B., Chow, W., Collins, J., Howe, K., Uliano-Silva, M., Fedrigo, O., Jarvis, E. D., Pérez-Tris, J., Illera, J. C., Liedvogel, M. (2022) Distinct patterns of genetic variation at low-recombining genomic regions represent haplotype structure. bioRxiv 2021.12.22.473882, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.12.22.473882 Li, H., & Ralph, P. (2019). Local PCA Shows How the Effect of Population Structure Differs Along the Genome. Genetics, 211 (1), 289–304. https://doi.org/10.1534/genetics.118.301747 Mérot, C., Berdan, E. L., Cayuela, H., Djambazian, H., Ferchaud, A.-L., Laporte, M., Normandeau, E., Ragoussis, J., Wellenreuther, M., & Bernatchez, L. (2021). Locally Adaptive Inversions Modulate Genetic Variation at Different Geographic Scales in a Seaweed Fly. Molecular Biology and Evolution, 38 (9), 3953–3971. https://doi.org/10.1093/molbev/msab143 | Distinct patterns of genetic variation at low-recombining genomic regions represent haplotype structure | Jun Ishigohoka, Karen Bascón-Cardozo, Andrea Bours, Janina Fuß, Arang Rhie, Jacquelyn Mountcastle, Bettina Haase, William Chow, Joanna Collins, Kerstin Howe, Marcela Uliano-Silva, Olivier Fedrigo, Erich D. Jarvis, Javier Pérez-Tris, Juan Carlos Il... | <p>Genetic variation of the entire genome represents population structure, yet individual loci can show distinct patterns. Such deviations identified through genome scans have often been attributed to effects of selection instead of randomness. Th... | | Genome Evolution, Molecular Evolution, Population Genetics / Genomics | Matteo Fumagalli | 2023-10-13 11:58:47 | ||
12 Jul 2017

Despite reproductive interference, the net outcome of reproductive interactions among spider mite species is not necessarily costlyThe pros and cons of mating with strangersRecommended by Vincent Calcagno based on reviews by Joël Meunier and Michael D Greenfield
Interspecific matings are by definition rare events in nature, but when they occur they can be very important, and not only because they might condition gene flow between species. Even when such matings have no genetic consequence, for instance if they do not yield any fertile hybrid offspring, they can still have an impact on the population dynamics of the species involved [1]. Such atypical pairings between heterospecific partners are usually regarded as detrimental or undesired; as they interfere with the occurrence or success of intraspecific matings, they are expected to cause a decline in absolute fitness. References [1] Gröning J. & Hochkirch A. 2008. Reproductive interference between animal species. The Quarterly Review of Biology 83: 257-282. doi: 10.1086/590510 [2] Clemente SH, Santos I, Ponce AR, Rodrigues LR, Varela SAM & Magalhaes S. 2017 Despite reproductive interference, the net outcome of reproductive interactions among spider mite species is not necessarily costly. bioRxiv 113274, ver. 4 of the 30th of June 2017. doi: 10.1101/113274 | Despite reproductive interference, the net outcome of reproductive interactions among spider mite species is not necessarily costly | Salomé H. Clemente, Inês Santos, Rita Ponce, Leonor R. Rodrigues, Susana A. M. Varela and Sara Magalhães | Reproductive interference is considered a strong ecological force, potentially leading to species exclusion. This supposes that the net effect of reproductive interactions is strongly negative for one of the species involved. Testing this requires... | | Behavior & Social Evolution, Evolutionary Ecology, Species interactions | Vincent Calcagno | 2017-03-06 11:48:08 | ||
24 Aug 2022

Density dependent environments can select for extremes of body sizeA population biological modeling approach for life history and body size evolutionRecommended by Wolf Blanckenhorn based on reviews by Frédéric Guillaume and 2 anonymous reviewersBody size evolution is a central theme in evolutionary biology. Particularly the question of when and how smaller body sizes can evolve continues to interest evolutionary ecologists, because most life history models, and the empirical evidence, document that large body size is favoured by natural and sexual selection in most (even small) organisms and environments at most times. How, then, can such a large range of body size and life history syndromes evolve and coexist in nature? The paper by Coulson et al. lifts this question to the level of the population, a relatively novel approach using so-called integral projection (simulation) models (IPMs) (as opposed to individual-based or game theoretical models). As is well outlined by (anonymous) Reviewer 1, and following earlier papers spearheading this approach in other life history contexts, the authors use the well-known carrying capacity (K) of population biology as the ultimate fitness parameter to be maximized or optimized (rather than body size per se), to ultimately identify factors and conditions promoting the evolution of extreme body sizes in nature. They vary (individual or population) size-structured growth trajectories to observe age and size at maturity, surivorship and fecundity/fertility schedules upon evaluating K (see their Fig. 1). Importantly, trade-offs are introduced via density-dependence, either for adult reproduction or for juvenile survival, in two (of several conceivable) basic scenarios (see their Table 2). All other relevant standard life history variables (see their Table 1) are assumed density-independent, held constant or zero (as e.g. the heritability of body size). The authors obtain evidence for disruptive selection on body size in both scenarios, with small size and a fast life history evolving below a threshold size at maturity (at the lowest K) and large size and a slow life history beyond this threshold (see their Fig. 2). Which strategy wins ultimately depends on the fitness benefits of delaying sexual maturity (at larger size and longer lifespan) at the adult stage relative to the preceeding juvenile mortality costs, in agreement with classic life history theory (Roff 1992, Stearns 1992). The modeling approach can be altered and refined to be applied to other key life history parameters and environments. These results can ultimately explain the evolution of smaller body sizes from large body sizes, or vice versa, and their corresponding life history syndromes, depending on the precise environmental circumstances. All reviewers agreed that the approach taken is technically sound (as far as it could be evaluated), and that the results are interesting and worthy of publication. In a first round of reviews various clarifications of the manuscript were suggested by the reviewers. The new version was substantially changed by the authors in response, to the extent that it now is a quite different but much clearer paper with a clear message palatable for the general reader. The writing is now to the point, the paper's focus becomes clear in the Introduction, Methods & Results are much less technical, the Figures illustrative, and the descriptions and interpretations in the Discussion are easy to follow. In general any reader may of course question the choice and realism of the scenarios and underlying assumptions chosen by the authors for simplicity and clarity, for instance no heritability of body size and no cost of reproduction (other than mortality). But this is always the case in modeling work, and the authors acknowledge and in fact suggest concrete extensions and expansions of their approach in the Discussion. References Coulson T., Felmy A., Potter T., Passoni G., Montgomery R.A., Gaillard J.-M., Hudson P.J., Travis J., Bassar R.D., Tuljapurkar S., Marshall D.J., Clegg S.M. (2022) Density-dependent environments can select for extremes of body size. bioRxiv, 2022.02.17.480952, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.02.17.480952 | Density dependent environments can select for extremes of body size | Tim Coulson, Anja Felmy, Tomos Potter, Gioele Passoni, Robert A Montgomery, Jean-Michel Gaillard, Peter J Hudson, Joseph Travis, Ronald D Bassar, Shripad D Tuljapurkar, Dustin Marshall, Sonya M Clegg | <p>Body size variation is an enigma. We do not understand why species achieve the sizes they do, and this means we also do not understand the circumstances under which gigantism or dwarfism is selected. We develop size-structured integral projecti... | | Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Theory, Life History | Wolf Blanckenhorn | 2022-02-21 07:59:04 | ||
13 Sep 2019

Deceptive combined effects of short allele dominance and stuttering: an example with Ixodes scapularis, the main vector of Lyme disease in the U.S.A.New curation method for microsatellite markers improves population genetics analysesRecommended by Aurelien Tellier based on reviews by Eric Petit, Martin Husemann and 2 anonymous reviewersGenetic markers are used for in modern population genetics/genomics to uncover the past neutral and selective history of population and species. Besides Single Nucleotide Polymorphisms (SNPs) obtained from whole genome data, microsatellites (or Short Tandem Repeats, SSR) have been common markers of choice in numerous population genetics studies of non-model species with large sample sizes [1]. Microsatellites can be used to uncover and draw inference of the past population demography (e.g. expansion, decline, bottlenecks…), population split, population structure and gene flow, but also life history traits and modes of reproduction (e.g. [2,3]). These markers are widely used in conservation genetics [4] or to study parasites or disease vectors [5]. Microsatellites do show higher mutation rate than SNPs increasing, on the one hand, the statistical power to infer recent events (for example crop domestication, [2,3]), while, on the other hand, decreasing their statistical power over longer time scales due to homoplasy [6]. References [1] Jarne, P., and Lagoda, P. J. (1996). Microsatellites, from molecules to populations and back. Trends in ecology & evolution, 11(10), 424-429. doi: 10.1016/0169-5347(96)10049-5 | Deceptive combined effects of short allele dominance and stuttering: an example with Ixodes scapularis, the main vector of Lyme disease in the U.S.A. | Thierry De Meeûs, Cynthia T. Chan, John M. Ludwig, Jean I. Tsao, Jaymin Patel, Jigar Bhagatwala, and Lorenza Beati | <p>Null alleles, short allele dominance (SAD), and stuttering increase the perceived relative inbreeding of individuals and subpopulations as measured by Wright’s FIS and FST. Ascertainment bias, due to such amplifying problems are usually caused ... | | Evolutionary Ecology, Other, Population Genetics / Genomics | Aurelien Tellier | 2019-05-02 20:52:08 |




