
Latest recommendations

| Id | Title | Authors▲ | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
05 Jan 2023

Promoting extinction or minimizing growth? The impact of treatment on trait trajectories in evolving populationsTrait trajectories in evolving populations: insights from mathematical modelsRecommended by Dominik Wodarz based on reviews by Rob Noble and 3 anonymous reviewersThe evolution of cells within organisms can be an important determinant of disease. This is especially clear in the emergence of tumors and cancers from the underlying healthy tissue. In the healthy state, homeostasis is maintained through complex regulatory processes that ensure a relatively constant population size of cells, which is required for tissue function. Tumor cells escape this homeostasis, resulting in uncontrolled growth and consequent disease. Disease progression is driven by further evolutionary processes within the tumor, and so is the response of tumors to therapies. Therefore, evolutionary biology is an important component required for a better understanding of carcinogenesis and the treatment of cancers. In particular, evolutionary theory helps define the principles of mutant evolution and thus to obtain a clearer picture of the determinants of tumor emergence and therapy responses. The study by Raatz and Traulsen [1] makes an important contribution in this respect. They use mathematical and computational models to investigate trait evolution in the context of evolutionary rescue, motivated by the dynamics of cancer, and also bacterial infections. This study views the establishment of tumors as cell dynamics in harsh environments, where the population is prone to extinction unless mutants emerge that increase evolutionary fitness, allowing them to expand (evolutionary rescue). The core processes of the model include growth, death, and mutations. Random mutations are assumed to give rise to cell lineages with different trait combinations, where the birth and death rates of cells can change. The resulting evolutionary trajectories are investigated in the models, and interesting new results were obtained. For example, the turnover of the population was identified as an important determinant of trait evolution. Turnover is defined as the balance between birth and death, with large rates corresponding to fast turnover and small rates to slow turnover. It was found that for fast cell turnover, a given adaptive step in the trait space results in a smaller increase in survival probability than for cell populations with slower turnover. In other words, evolutionary rescue is more difficult to achieve for fast compared to slow turnover populations. While more mutants can be produced for faster cell turnover rates, the analysis showed that this is not sufficient to overcome the barrier to the evolutionary rescue. This result implies that aggressive tumors with fast cell birth and death rates are less likely to persist and progress than tumors with lower turnover rates. This work emphasizes the importance of measuring the turnover rate in different tumors to advance our understanding of the determinants of tumor initiation and progression. The authors discuss that the well-documented heterogeneity in tumors likely also applies to cellular turnover. If a tumor consists of sub-populations with faster and slower turnover, it is possible that a slower turnover cell clone (e.g. characterized by a degree of dormancy) would enjoy a selective advantage. Another source of heterogeneity in turnover could be given by the hierarchical organization of tumors. Similar to the underlying healthy tissue, many tumors are thought to be maintained by a population of cancer stem cells, while the tumor bulk is made up of more differentiated cells. Tissue stem cells tend to be characterized by a lower turnover than progenitor or transit-amplifying cells. Depending on the assumptions about the self-renewal capacity of these different cell populations, the potential for evolutionary rescue could be different depending on the cell compartment in which the mutant emerges. This might be interesting to explore in the future. There are also implications for treatment. Two types of treatment were investigated: density-affecting treatments in which the density of cells is reduced without altering their trait parameters, and trait-affecting treatments in which the birth and/or death rates are altered. Both types of treatment were found to change the trajectories of trait adaptation, which has potentially important practical implications. Interestingly, it was found that competitive release during treatment can result in situations where after treatment cessation, the non-extinct populations recover to reach sizes that were higher than in the absence of treatment. This points towards the potential of adaptive therapy approaches, where sensitive cells are maintained to some extent to suppress resistant clones [2] competitively. In this context, it is interesting that the success of such approaches might also depend on the turnover of the tumor cell population, as shown by a recent mathematical modeling study [3]. In particular, it was found that adaptive therapy is less likely to work for slow compared to fast turnover tumors. Yet, the current study by Raatz and Traulsen [1] suggests that tumors are more likely to evolve in a slow turnover setting. While there is strong relevance of this analysis for tumor evolution, the results generated in this study have more general relevance. Besides tumors, the paper discusses applications to bacterial disease dynamics in some detail, which is also interesting to compare and contrast to evolutionary processes in cancer. Overall, this study provides insights into the dynamics of evolutionary rescue that represent valuable additions to evolutionary theory. References [1] Raatz M, Traulsen A (2023) Promoting extinction or minimizing growth? The impact of treatment on trait trajectories in evolving populations. bioRxiv, 2022.06.17.496570, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.06.17.496570 [2] Gatenby RA, Silva AS, Gillies RJ, Frieden BR (2009) Adaptive Therapy. Cancer Research, 69, 4894–4903. https://doi.org/10.1158/0008-5472.CAN-08-3658 [3] Strobl MAR, West J, Viossat Y, Damaghi M, Robertson-Tessi M, Brown JS, Gatenby RA, Maini PK, Anderson ARA (2021) Turnover Modulates the Need for a Cost of Resistance in Adaptive Therapy. Cancer Research, 81, 1135–1147. https://doi.org/10.1158/0008-5472.CAN-20-0806 | Promoting extinction or minimizing growth? The impact of treatment on trait trajectories in evolving populations | Michael Raatz, Arne Traulsen | <p style="text-align: justify;">When cancers or bacterial infections establish, small populations of cells have to free themselves from homoeostatic regulations that prevent their expansion. Trait evolution allows these populations to evade this r... | | Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Theory | Dominik Wodarz | 2022-06-18 08:44:37 | ||
04 Jul 2022
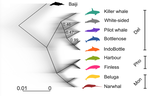
A genomic assessment of the marine-speciation paradox within the toothed whale superfamily DelphinoideaReticulated evolution marks the rapid diversification of the DelphinoidaeRecommended by Michael C. Fontaine based on reviews by Christelle Fraïsse, Simon Henry Martin, Andrew Foote and 2 anonymous reviewersHistorically neglected or considered a rare aberration in animals under the biological species concept, interspecific hybridisation has by now been recognised to be taxonomically widespread, particularly in rapidly diversifying groups (Dagilis et al. 2021; Edelman & Mallet 2021; Mallet et al. 2016; Seehausen 2004). Yet the prevalence of introgressive hybridizations, its evolutionary significance, and its impact on species diversification remain a hot topic of research in evolutionary biology. The rapid increase in genomic resources now available for non-model species has significantly contributed to the detection of introgressive hybridization across taxa showing that reticulated evolution is far more common in the animal kingdom than historically considered. Yet, detecting it, quantifying its magnitude, and assessing its evolutionary significance remains a challenging endeavour with constantly evolving methodologies to better explore and exploit genomic data (Blair & Ané 2020; Degnan & Rosenberg 2009; Edelman & Mallet 2021; Hibbins & Hahn 2022). In the marine realm, the dearth of geographic barriers and the large dispersal abilities of pelagic species like cetaceans have raised the questions of how populations and species can diverge and adapt to distinct ecological conditions in face of potentially large gene-flow, the so-called marine speciation paradox (Bierne et al. 2003). Contemporaneous hybridization among cetacean species has been widely documented in nature despite large phenotypic differences (Crossman et al. 2016). The historical prevalence of reticulated evolution, its evolutionary significance, and how it might have impacted the evolutionary history and diversification of the cetaceans have however remained elusive so far. Recent phylogenomic studies suggested that introgression has been prevalent in cetacean evolutionary history with instances reported among baleen whales (mysticetes) (Árnason et al. 2018) and among toothed whales (odontocetes), especially in the rapidly diversifying dolphins family of the Delphininae (Guo et al. 2021; Moura et al. 2020). Analysing publicly available whole-genome data from nine cetacean species across three families of Delphinoidae – dolphins, porpoises, and monondontidae – using phylogenomics and demo-genetics approaches, Westbury and colleagues (2022) take a step further in documenting that evolution among these species has been far from a simple bifurcating tree. Instead, their study describes widespread occurrences of introgression among Delphinoidae, drawing a complex picture of reticulated evolutionary history. After describing major topology discordance in phylogenetic gene trees along the genome, the authors use a panel of approaches to disentangle introgression from incomplete lineage sorting (ILS), the two most common causes of tree topology discordances (Hibbins & Hahn 2022). Applying popular tests that separate introgression from ILS, such as the Patterson’s D (a.k.a. ABBA-BABA) test (Durand et al. 2011; Green et al. 2010), QuIBL (Edelman et al. 2019), and D-FOIL (Pease & Hahn 2015), the authors report that signals of introgression are present in the genomes of most (if not all) the cetacean species included in their study. However, this picture needs to be nuanced. Most introgression signals seem to echo old introgression events that occurred primarily among ancestors. This is suggested by the differential signals of topology discordance along the genome when considering sliding windows along the genome of varying sizes (50kb, 100kb, and 1Mb), and by patterns of excess derived allele sharing along branches of the species tree, as captured by the f-branch test (Malinsky et al. 2021; Malinsky et al. 2018). The authors further investigated the dynamic of cessation of gene flow (and/or ILS) between species using the F1 hybrid PSMC (or hPSMC) approach (Cahill et al. 2016). By estimating the cross-coalescent rates (CRR) between species pairs with time in light of previously estimated species divergence times (McGowen et al. 2020), the authors report that gene flow (and/or ILS) most likely has stopped by now among most species, but it may have lasted for more than half of the time since the species split from each other. According to the author, this result may reflect the slow process by which reproductive isolation would have evolved between cetacean lineages, and that species isolation was marked by significant introgression events. Now, while the present study provides a good overview of how complex is the reticulated evolutionary history of the Delphinoidae, getting a complete picture will require overcoming a few important limitations. The first ones are methodological and related to the phylogenomic analyses. Given the sampling design with one diploid genome per species, the authors could not phase the data into the parental haplotypes, but instead relied on a majority consensus creating mosaic haploidized genomes representing a mixture between the two parental copies. Moreover, by using large genomic windows (≥50kb) that likely span multiple independent loci, phylogenetic analyses in windows encompassed distinct phylogenetic signals, potentially leading to bias and inaccuracy in the inferences. Thawornwattana et al (2018) previously showed that this “concatenation approach” could significantly impact phylogenetic inferences. They proposed instead to use loci small enough to minimise the risk of intra-locus recombination and to consider them in blocks of non-recombining loci along the genome in order to conduct phylogenetic analysed, ideally under the multi-species coalescent (MSC) that can account for ILS (e.g. BPP; Flouri et al. 2018; Jiao et al. 2020; Yang 2015). Such an approach applied to the diversification of the Delphinidae may reveal substantial changes compared to the currently admitted species tree. Inaccuracy in the species tree estimation may have a major impact on the introgression analyses conducted in this study since the species tree and branching order must be supplied in the introgression analyses to properly disentangle introgression from ILS. Here, the authors rely on the tree topology that was previously estimated in McGowen et al. (2020), which they also recovered using their consensus estimation from ASTRAL-III (Zhang et al. 2018). While the methodologies accounted to a certain extent for ILS, albeit with potential bias induced by the concatenation approach, they ignore the presumably large amount of introgression among species during the diversification process. Estimating species branching order while ignoring introgression can lead to major bias in the phylogenetic inference and can lead to incorrect topologies. Even if these MSC-based methods account for ILS, inferences can become very inaccurate or even break down as gene flow increases (see for ex. Jiao et al. 2020; Müller et al. in press; Solís-Lemus et al. 2016). Dedicated approaches have been developed to model explicitly introgression together with ILS to estimate phylogenetic networks (Blair & Ané 2020; Rabier et al. 2021) or in isolation-with-migration model (Müller et al. in press; Wang et al. 2020). Future studies revisiting the reticulated evolutionary history of the Delphinoidae with such approaches may not only precise the species branching order, but also deliver a finer view of the magnitude and prevalence of introgression during the evolutionary history of these species. A final part of Westbury et al. (2022)'s study set out to test whether historical periods of low abundance could have facilitated hybridization among Delphinoidae species. During these periods of low abundance, species may encounter only a limited number of conspecifics and may consider individuals from other species as suitable mating partners, leading to hybridisation (Crossman et al. 2016; Edwards et al. 2011; Westbury et al. 2019). The authors tested this hypothesis by considering genome-wide genetic diversity (or heterozygosity) as a proxy of historical effective population size (Ne), itself as a proxy of the evolution of census size with time. They also try to link historical Ne variation (from PSMC, Li & Durbin 2011) with their estimated time to cessation of gene flow or ILS (from the CRR of hPSMC). However, no straightforward relationship was found between the genetic diversity and the propensity of species to hybridize, nor was there any clear link between Ne variation through time and the cessation of gene flow or ILS. Such a lack of relationship may not come as a surprise, since the determinants of genome-wide genetic diversity and its variation through evolutionary time-scale are far more diverse and complex than just a direct link with hybridization, introgression, or even with the census population size. In fact, genetic diversity results from the balance between all the evolutionary processes at play in the species' evolutionary history (see the review of Ellegren & Galtier 2016). Other important factors can strongly impact genetic diversity, including demography and structure, but also linked selection (Boitard et al. 2022; Buffalo 2021; Ellegren & Galtier 2016). All in all, Westbury and coll. (2022) present here an interesting study providing an additional step towards resolving and understanding the complex evolutionary history of the Delphinoidae, and shedding light on the importance of introgression during the diversification of these cetacean species. Prospective work improving upon the taxonomic sampling, with additional genomic data for each species, considered with dedicated approaches tailored at estimating species tree or network while accounting for ILS and introgression will be key for refining the picture depicted in this study. Down the road, altogether these studies will contribute to assessing the evolutionary significance of introgression on the diversification of Delphinoides, and more generally on the diversification of cetacean species, which still remain an open and exciting perspective. References Árnason Ú, Lammers F, Kumar V, Nilsson MA, Janke A (2018) Whole-genome sequencing of the blue whale and other rorquals finds signatures for introgressive gene flow. Science Advances 4, eaap9873. https://doi.org/10.1126/sciadv.aap9873 Bierne N, Bonhomme F, David P (2003) Habitat preference and the marine-speciation paradox. Proceedings of the Royal Society of London. Series B: Biological Sciences 270, 1399-1406. https://doi.org/10.1098/rspb.2003.2404 Blair C, Ané C (2020) Phylogenetic Trees and Networks Can Serve as Powerful and Complementary Approaches for Analysis of Genomic Data. Systematic Biology 69, 593-601. https://doi.org/10.1093/sysbio/syz056 Boitard S, Arredondo A, Chikhi L, Mazet O (2022) Heterogeneity in effective size across the genome: effects on the inverse instantaneous coalescence rate (IICR) and implications for demographic inference under linked selection. Genetics 220, iyac008. https://doi.org/10.1093/genetics/iyac008 Buffalo V (2021) Quantifying the relationship between genetic diversity and population size suggests natural selection cannot explain Lewontin's Paradox. e-Life 10, e67509. https://doi.org/10.7554/eLife.67509 Cahill JA, Soares AE, Green RE, Shapiro B (2016) Inferring species divergence times using pairwise sequential Markovian coalescent modelling and low-coverage genomic data. Philos Trans R Soc Lond B Biol Sci 371, 20150138. https://doi.org/10.1098/rstb.2015.0138 Crossman CA, Taylor EB, Barrett‐Lennard LG (2016) Hybridization in the Cetacea: widespread occurrence and associated morphological, behavioral, and ecological factors. Ecology and Evolution 6, 1293-1303. https://doi.org/10.1002/ece3.1913 Dagilis AJ, Peede D, Coughlan JM, Jofre GI, D’Agostino ERR, Mavengere H, Tate AD, Matute DR (2021) 15 years of introgression studies: quantifying gene flow across Eukaryotes. biorXiv, 2021.1106.1115.448399. https://doi.org/10.1101/2021.06.15.448399 Degnan JH, Rosenberg NA (2009) Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol Evol 24, 332-340. https://doi.org/10.1016/j.tree.2009.01.009 Durand EY, Patterson N, Reich D, Slatkin M (2011) Testing for ancient admixture between closely related populations. Mol Biol Evol 28, 2239-2252. https://doi.org/10.1093/molbev/msr048 Edelman NB, Frandsen PB, Miyagi M, Clavijo B, Davey J, Dikow RB, Garcia-Accinelli G, Van Belleghem SM, Patterson N, Neafsey DE, Challis R, Kumar S, Moreira GRP, Salazar C, Chouteau M, Counterman BA, Papa R, Blaxter M, Reed RD, Dasmahapatra KK, Kronforst M, Joron M, Jiggins CD, McMillan WO, Di Palma F, Blumberg AJ, Wakeley J, Jaffe D, Mallet J (2019) Genomic architecture and introgression shape a butterfly radiation. Science 366, 594-599. https://doi.org/10.1126/science.aaw2090 Edelman NB, Mallet J (2021) Prevalence and Adaptive Impact of Introgression. Annual Review of Genetics 55, 265-283. https://doi.org/10.1146/annurev-genet-021821-020805 Edwards CJ, Suchard MA, Lemey P, Welch JJ, Barnes I, Fulton TL, Barnett R, O'Connell TC, Coxon P, Monaghan N, Valdiosera CE, Lorenzen ED, Willerslev E, Baryshnikov GF, Rambaut A, Thomas MG, Bradley DG, Shapiro B (2011) Ancient hybridization and an Irish origin for the modern polar bear matriline. Curr Biol 21, 1251-1258. https://doi.org/10.1016/j.cub.2011.05.058 Ellegren H, Galtier N (2016) Determinants of genetic diversity. Nat Rev Genet 17, 422-433. https://doi.org/10.1038/nrg.2016.58 Flouri T, Jiao X, Rannala B, Yang Z (2018) Species Tree Inference with BPP Using Genomic Sequences and the Multispecies Coalescent. Mol Biol Evol 35, 2585-2593. https://doi.org/10.1093/molbev/msy147 Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, Hansen NF, Durand EY, Malaspinas AS, Jensen JD, Marques-Bonet T, Alkan C, Prufer K, Meyer M, Burbano HA, Good JM, Schultz R, Aximu-Petri A, Butthof A, Hober B, Hoffner B, Siegemund M, Weihmann A, Nusbaum C, Lander ES, Russ C, Novod N, Affourtit J, Egholm M, Verna C, Rudan P, Brajkovic D, Kucan Z, Gusic I, Doronichev VB, Golovanova LV, Lalueza-Fox C, de la Rasilla M, Fortea J, Rosas A, Schmitz RW, Johnson PLF, Eichler EE, Falush D, Birney E, Mullikin JC, Slatkin M, Nielsen R, Kelso J, Lachmann M, Reich D, Paabo S (2010) A draft sequence of the Neandertal genome. Science 328, 710-722. https://doi.org/10.1126/science.1188021 Guo W, Sun D, Cao Y, Xiao L, Huang X, Ren W, Xu S, Yang G (2021) Extensive Interspecific Gene Flow Shaped Complex Evolutionary History and Underestimated Species Diversity in Rapidly Radiated Dolphins. Journal of Mammalian Evolution 29, 353-367. https://doi.org/10.1007/s10914-021-09581-6 Hibbins MS, Hahn MW (2022) Phylogenomic approaches to detecting and characterizing introgression. Genetics 220, iyab173. https://doi.org/10.1093/genetics/iyab173 Jiao X, Flouri T, Rannala B, Yang Z (2020) The Impact of Cross-Species Gene Flow on Species Tree Estimation. Syst Biol 69, 830-847. https://doi.org/10.1093/sysbio/syaa001 Li H, Durbin R (2011) Inference of human population history from individual whole-genome sequences. Nature 475, 493-496. https://doi.org/10.1038/nature10231 Malinsky M, Matschiner M, Svardal H (2021) Dsuite - Fast D-statistics and related admixture evidence from VCF files. Mol Ecol Resour 21, 584-595. https://doi.org/10.1111/1755-0998.13265 Malinsky M, Svardal H, Tyers AM, Miska EA, Genner MJ, Turner GF, Durbin R (2018) Whole-genome sequences of Malawi cichlids reveal multiple radiations interconnected by gene flow. Nature Ecology & Evolution 2, 1940-1955. https://doi.org/10.1038/s41559-018-0717-x Mallet J, Besansky N, Hahn MW (2016) How reticulated are species? Bioessays 38, 140-149. https://doi.org/10.1002/bies.201500149 McGowen MR, Tsagkogeorga G, Alvarez-Carretero S, Dos Reis M, Struebig M, Deaville R, Jepson PD, Jarman S, Polanowski A, Morin PA, Rossiter SJ (2020) Phylogenomic Resolution of the Cetacean Tree of Life Using Target Sequence Capture. Syst Biol 69, 479-501. https://doi.org/10.1093/sysbio/syz068 Moura AE, Shreves K, Pilot M, Andrews KR, Moore DM, Kishida T, Möller L, Natoli A, Gaspari S, McGowen M, Chen I, Gray H, Gore M, Culloch RM, Kiani MS, Willson MS, Bulushi A, Collins T, Baldwin R, Willson A, Minton G, Ponnampalam L, Hoelzel AR (2020) Phylogenomics of the genus Tursiops and closely related Delphininae reveals extensive reticulation among lineages and provides inference about eco-evolutionary drivers. Molecular Phylogenetics and Evolution 146,107047. https://doi.org/10.1016/j.ympev.2020.106756 Müller NF, Ogilvie HA, Zhang C, Fontaine MC, Amaya-Romero JE, Drummond AJ, Stadler T (in press) Joint inference of species histories and gene flow. Syst Biol. Pease JB, Hahn MW (2015) Detection and Polarization of Introgression in a Five-Taxon Phylogeny. Syst Biol 64, 651-662. https://doi.org/10.1093/sysbio/syv023 Rabier CE, Berry V, Stoltz M, Santos JD, Wang W, Glaszmann JC, Pardi F, Scornavacca C (2021) On the inference of complex phylogenetic networks by Markov Chain Monte-Carlo. PLoS Comput Biol 17, e1008380. https://doi.org/10.1371/journal.pcbi.1008380 Seehausen O (2004) Hybridization and adaptive radiation. Trends Ecol Evol 19, 198-207. https://doi.org/10.1016/j.tree.2004.01.003 Solís-Lemus C, Yang M, Ané C (2016) Inconsistency of Species Tree Methods under Gene Flow. Syst Biol 65, 843-851. https://doi.org/10.1093/sysbio/syw030 Thawornwattana Y, Dalquen D, Yang Z, Tamura K (2018) Coalescent Analysis of Phylogenomic Data Confidently Resolves the Species Relationships in the Anopheles gambiae Species Complex. Molecular Biology and Evolution 35, 2512-2527. https://doi.org/10.1093/molbev/msy158 Wang K, Mathieson I, O’Connell J, Schiffels S (2020) Tracking human population structure through time from whole genome sequences. PLOS Genetics 16, e1008552. https://doi.org/10.1371/journal.pgen.1008552 Westbury MV, Cabrera AA, Rey-Iglesia A, Cahsan BD, Duchêne DA, Hartmann S, Lorenzen ED (2022) A genomic assessment of the marine-speciation paradox within the toothed whale superfamily Delphinoidea. bioRxiv, 2020.10.23.352286, ver. 7 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.10.23.352286 Westbury MV, Petersen B, Lorenzen ED (2019) Genomic analyses reveal an absence of contemporary introgressive admixture between fin whales and blue whales, despite known hybrids. PLoS ONE 14, e0222004. https://doi.org/10.1371/journal.pone.0222004 Yang Z (2015) The BPP program for species tree estimation and species delimitation. Current Zoology 61, 854-865. https://doi.org/10.1093/czoolo/61.5.854 Zhang C, Rabiee M, Sayyari E, Mirarab S (2018) ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19, 153. https://doi.org/10.1186/s12859-018-2129-y | A genomic assessment of the marine-speciation paradox within the toothed whale superfamily Delphinoidea | Michael V Westbury, Andrea A Cabrera, Alba Rey-Iglesia, Binia De Cahsan, David A. Duchêne, Stefanie Hartmann, Eline D Lorenzen | <p>The importance of post-divergence gene flow in speciation has been documented across a range of taxa in recent years, and may have been especially widespread in highly mobile, wide-ranging marine species, such as cetaceans. Here, we studied ind... | | Evolutionary Dynamics, Hybridization / Introgression, Molecular Evolution, Phylogenetics / Phylogenomics, Speciation | Michael C. Fontaine | 2020-10-25 08:55:50 | ||
22 Feb 2023
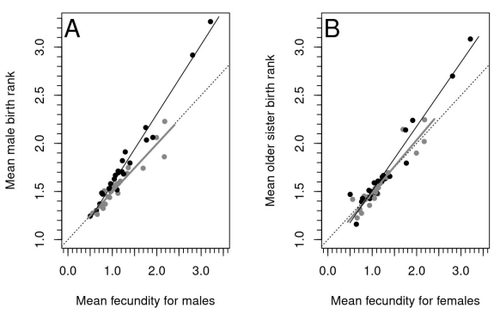
Increased birth rank of homosexual males: disentangling the older brother effect and sexual antagonism hypothesisEvolutionary or proximal explanations for human male homosexual mate preference?Recommended by Jacqui A. Shykoff based on reviews by Ray Blanchard and 1 anonymous reviewer based on reviews by Ray Blanchard and 1 anonymous reviewer
Natural populations do not consist of only perfectly adapted individuals. If they did, of course, there would be no fodder for evolution by natural selection. And natural selection is operating all the time, winnowing out less well adapted phenotypes through differential reproduction and survival. Demonstrations of natural selection modifying characters-state distributions to bring phenotypes closer to their optima abound in the evolution literature, with examples of short- and long-term changes in phenotype and allele frequencies. However, evolutionary biologists know that populations cannot reach their adaptive peaks. Natural selection is tracking a moving target, always with some generations of lag time. The adaptive landscape is multidimensional, so the optimal combination of multiple character states may be impossible because of constraints and trade-offs. Natural selection does not operate alone or in isolation – new mutations and migrants that were selected under other conditions will inject locally non-adaptive genetic variation and genetic drift can change allele frequencies in random directions. We understand these processes that generate and maintain less advantageous variants on a continuous gradient from an optimal phenotype in a fitness landscape. More puzzling are heritable polymorphisms with distinct morphologies, physiologies or behaviours maintained in populations despite their measurably lower reproductive success. But a complete model of evolution must also be able to accommodate these Darwinian paradoxes. Raymond et al. (2023) investigate one such Darwinian paradox: In humans, male homosexual mate preference is heritable and is associated with a large reduction in offspring production but nonetheless occurs at relatively high frequencies in most human populations. Furthermore, multiple studies have found that homosexual men come from families that are, on average, larger than those of heterosexual men and that homosexual men have, on average, higher birth rank than do heterosexual men, i.e., having more older siblings and, particularly, more older brothers. Two types of mechanisms consistent with these observations have been proposed: 1) An evolutionary mechanism of sex-antagonistic pleiotropy, whereby highly fecund mothers are more likely to produce homosexual sons, and 2) A mechanistic explanation whereby successive male pregnancies alter the uterine environment by increasing the probability of an immune reaction by the mother to her male fetus, altering development of sexually dimorphic brain structures relevant to sexual orientation. In this article, the authors explore these two mechanisms of sex-antagonistic effects (AE) and fraternal birth order effects (FBOE) and test how well they account for patterns of male homosexuality in population and family data. Clearly, these two effects are somewhat confounded because high birth ranks can only occur in large families. If, indeed, the probability of male homosexuality increases with increasing numbers of (maternal) older brothers, homosexual males will be more common in larger families. Similarly, if high female fecundity leads to a higher probability of male homosexuality via sex-antagonistic effects, homosexual males will, on average, have more older brothers. To disentangle the actions of these two effects the authors modelled the relationship between birth rank and population fecundity and investigated whether AE or FBOE modified this relationship for homosexual men. Simulation results were compared with aggregated population data from 13 countries. Family data on individuals’ sexual preference, birth rank and number of male and female siblings from France, Greece and Indonesia were analysed with generalised linear models and Bayesian approaches to test for a signal of AE or FBOE. These analyses revealed a significant older-brother effect (FBOE) explaining patterns of occurrence of homosexuality in population and family data but no significant independent sex-antagonistic effect (AE). Thus larger family sizes of homosexual men appear due to the older-brother effect, with individuals of high birth rank coming necessarily from large sibships. The simulation approach also revealed that modelling a fraternal birth order effect (FBOE), such that individuals with more older brothers are more likely to be homosexual, generates an artefactual older sister effect simply because homosexual men are overrepresented at higher birth ranks. Older-sister effects reported in the literature may, therefore, be statistical artefacts of an underlying older-brother effect. This paper is interesting for a number of reasons. It does an excellent job of explaining, identifying and dealing with estimation biases and testing for artefactual relationships generated by collinearity. It applies state-of-the art analytical/statistical tools. It breaks down two colinear effects and shows that only one really explains phenotypic variation. This is a great example of how to disentangle correlated variables that may or may not both contribute to trait variation. But most intriguingly, we are left without evidence for an evolutionary mechanism that compensates the large fitness cost associated with male homosexuality in humans. How can we explain high heritability maintained in the face of strong directional selection that should erode heritable genetic variation? The usual suspects include cryptic compensatory mechanisms yet to be discovered or flawed estimates of selection or heritability. For example, data on heritability of male homosexual mate preference in humans come from twin studies and twins share birth rank as well as alleles. Thus it is possible that heritability is over-estimated, including the environmental component associated with birth rank. If, as the authors demonstrate here, birth rank is the strongest predictor of male homosexual mate preference, selection may be acting on a non-heritable plastic component of phenotypic variation. This could explain why heritable variation is not exhausted by selection, rendering the paradox less paradoxical, but fails to provide an adaptive explanation for the maintenance of male homosexual mate preference. References Raymond M., Turek D., Durand V., Nila S., Suryobroto B., Vadez J., Barthes J., Apostolou M. and Crochet P.-A. (2023) Increased birth rank of homosexual males: disentangling the older brother effect and sexual antagonism hypothesis. bioRxiv, 2022.02.22.481477, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.02.22.481477 | Increased birth rank of homosexual males: disentangling the older brother effect and sexual antagonism hypothesis | Michel Raymond, Daniel Turek, Valerie Durand, Sarah Nila, Bambang Suryobroto, Julien Vadez, Julien Barthes, Menelaos Apostolou, Pierre-André Crochet | <p style="text-align: justify;">Male homosexual orientation remains a Darwinian paradox, as there is no consensus on its evolutionary (ultimate) determinants. One intriguing feature of homosexual men is their higher male birth rank compared to het... | | Life History, Other, Phenotypic Plasticity, Reproduction and Sex | Jacqui A. Shykoff | 2022-03-03 11:28:44 | ||
23 Nov 2020

Wolbachia and host intrinsic reproductive barriers contribute additively to post-mating isolation in spider mitesSpeciation in spider mites: disentangling the roles of Wolbachia-induced vs. nuclear mating incompatibilitiesRecommended by Jan Engelstaedter based on reviews by Wolfgang Miller and 1 anonymous reviewerCytoplasmic incompatibility (CI) is a mating incompatibility that is induced by maternally inherited endosymbionts in many arthropods. These endosymbionts include, most famously, the alpha-proteobacterium Wolbachia pipientis (Yen & Barr 1971; Werren et al. 2008) but also the Bacteroidetes bacterium Cardinium hertigii (Zchori-Fein et al. 2001), a gamma-proteobacterium of the genus Rickettsiella (Rosenwald et al. 2020) and another, as yet undescribed alpha-proteobacterium (Takano et al. 2017). CI manifests as embryonic mortality in crosses between infected males and females that are uninfected or infected with a different strain, whereas embryos develop normally in all other crosses. This phenotype may enable the endosymbionts to spread rapidly within their host population. Exploiting this, CI-inducing Wolbachia are being harnessed to control insect-borne diseases (e.g., O'Neill 2018). Much progress elucidating the genetic basis and developmental mechanism of CI has been made in recent years, but many open questions remain (Shropshire et al. 2020). References Bordenstein, S. R., O'Hara, F. P., and Werren, J. H. (2001). Wolbachia-induced incompatibility precedes other hybrid incompatibilities in Nasonia. Nature, 409(6821), 707-710. doi: https://doi.org/10.1038/35055543 | Wolbachia and host intrinsic reproductive barriers contribute additively to post-mating isolation in spider mites | Miguel A. Cruz, Sara Magalhães, Élio Sucena, Flore Zélé | <p>Wolbachia are widespread maternally-inherited bacteria suggested to play a role in arthropod host speciation through induction of cytoplasmic incompatibility, but this hypothesis remains controversial. Most studies addressing Wolbachia-induced ... | | Evolutionary Ecology, Hybridization / Introgression, Life History, Reproduction and Sex, Speciation, Species interactions | Jan Engelstaedter | 2020-07-09 10:18:28 | ||
26 Oct 2020
Power and limits of selection genome scans on temporal data from a selfing populationDetecting loci under natural selection from temporal genomic data of selfing populationsRecommended by Matteo Fumagalli based on reviews by Christian Huber and 2 anonymous reviewers
The observed levels of genomic diversity in contemporary populations are the result of changes imposed by several evolutionary processes. Among them, natural selection is known to dramatically shape the genetic diversity of loci associated with phenotypes which affect the fitness of carriers. As such, many efforts have been dedicated towards developing methods to detect signatures of natural selection from genomes of contemporary samples [1]. References [1] Stern AJ, Nielsen R (2019) Detecting Natural Selection. In: Handbook of Statistical Genomics , pp. 397–40. John Wiley and Sons, Ltd. https://doi.org/10.1002/9781119487845.ch14 | Power and limits of selection genome scans on temporal data from a selfing population | Miguel Navascués, Arnaud Becheler, Laurène Gay, Joëlle Ronfort, Karine Loridon, Renaud Vitalis | <p>Tracking genetic changes of populations through time allows a more direct study of the evolutionary processes acting on the population than a single contemporary sample. Several statistical methods have been developed to characterize the demogr... | | Adaptation, Bioinformatics & Computational Biology, Population Genetics / Genomics, Reproduction and Sex | Matteo Fumagalli | 2020-05-08 10:34:31 | ||
22 May 2017
Can Ebola Virus evolve to be less virulent in humans?A new hypothesis to explain Ebola's high virulenceRecommended by Virginie Ravigné and François Blanquart based on reviews by Virginie Ravigné and François Blanquart
The tragic 2014-2016 Ebola outbreak that resulted in more than 28,000 cases and 11,000 deaths in West Africa [1] has been a surprise to the scientific community. Before 2013, the Ebola virus (EBOV) was known to produce recurrent outbreaks in remote villages near tropical rainforests in Central Africa, never exceeding a few hundred cases with very high virulence. Both EBOV’s ability to circulate for several months in large urban human populations and its important mutation rate suggest that EBOV’s virulence could evolve and to some extent adapt to human hosts [2]. Up to now, the high virulence of EBOV in humans was generally thought to be maladaptive, the virus being adapted to circulating in wild animal populations (e.g. fruit bats [3]). As a logical consequence, EBOV virulence could be expected to decrease during long epidemics in humans. The present paper by Sofonea et al. [4] challenges this view and explores how, given EBOV’s life cycle and known epidemiological parameters, virulence is expected to evolve in the human host during long epidemics. The main finding of the paper is that there is no chance that EBOV’s virulence decreases in the short and long terms. The main underlying mechanism is that EBOV is also transmitted by dead bodies, which limits the cost of virulence. In itself the idea that selection should select for higher virulence in diseases that are also transmitted after host death will sound intuitive for most evolutionary epidemiologists. The accomplishment of the paper is to make a very strong case that the parameter range where virulence could decrease is very small. The paper further provides scientifically grounded arguments in favor of the safe management of corpses. Safe burial of corpses is culturally difficult to impose. The present paper shows that in addition to instantaneously decreasing the spread of the virus, safe burial may limit virulence increase in the short term and favor of less virulent strains in the long term. Altogether these results make a timely and important contribution to the knowledge and understanding of EBOV. References [1] World Health Organization. 2016. WHO: Ebola situation report - 10 June 2016. [2] Kupferschmidt K. 2014. Imagining Ebola’s next move. Science 346: 151–152. doi: 10.1126/science.346.6206.151 [3] Leroy EM, Kumulungui B, Pourrut X, Rouquet P, Hassanin A, Yaba P, Délicat A, Paweska, Gonzalez JP and Swanepoel R. 2005. Fruit bats as reservoirs of Ebola virus. Nature 438: 575–576. doi: 10.1038/438575a [4] Sofonea MT, Aldakak L, Boullosa LFVV and Alizon S. 2017. Can Ebola Virus evolve to be less virulent in humans? bioRxiv 108589, ver. 3 of 19th May 2017; doi: 10.1101/108589 | Can Ebola Virus evolve to be less virulent in humans? | Mircea T. Sofonea, Lafi Aldakak, Luis Fernando Boullosa, Samuel Alizon | Understanding Ebola Virus (EBOV) virulence evolution is not only timely but also raises specific questions because it causes one pf the most virulent human infections and it is capable of transmission after the death of its host. Using a compartme... | | Evolutionary Epidemiology | Virginie Ravigné | 2017-02-15 13:25:58 | ||
30 Aug 2021
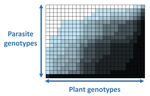
The quasi-universality of nestedness in the structure of quantitative plant-parasite interactionsNestedness and modularity in plant-parasite infection networksRecommended by Santiago Elena based on reviews by Rubén González and 2 anonymous reviewers
In a landmark paper, Flores et al. (2011) showed that the interactions between bacteria and their viruses could be nicely described using a bipartite infection networks. Two quantitative properties of these networks were of particular interest, namely modularity and nestedness. Modularity emerges when groups of host species (or genotypes) shared groups of viruses. Nestedness provided a view of the degree of specialization of both partners: high nestedness suggests that hosts differ in their susceptibility to infection, with some highly susceptible host genotypes selecting for very specialized viruses while strongly resistant host genotypes select for generalist viruses. Translated to the plant pathology parlance, this extreme case would be equivalent to a gene-for-gene infection model (Flor 1956): new mutations confer hosts with resistance to recently evolved viruses while maintaining resistance to past viruses. Likewise, virus mutations for expanding host range evolve without losing the ability to infect ancestral host genotypes. By contrast, a non-nested network would represent a matching-allele infection model (Frank 2000) in which each interacting organism evolves by losing its capacity to resist/infect its ancestral partners, resembling a Red Queen dynamic. Obviously, the reality is more complex and may lie anywhere between these two extreme situations. Recently, Valverde et al. (2020) developed a model to explain the emergence of nestedness and modularity in plant-virus infection networks across diverse habitats. They found that local modularity could coexist with global nestedness and that intraspecific competition was the main driver of the evolution of ecosystems in a continuum between nested-modular and nested networks. These predictions were tested with field data showing the association between plant host species and different viruses in different agroecosystems (Valverde et al. 2020). The effect of interspecific competition in the structure of empirical plant host-virus infection networks was also tested by McLeish et al. (2019). Besides data from agroecosystems, evolution experiments have also shown the pervasive emergence of nestedness during the diversification of independently-evolved lineages of potyviruses in Arabidopsis thaliana genotypes that differ in their susceptibility to infection (Hillung et al. 2014; González et al. 2019; Navarro et al. 2020). In their study, Moury et al. (2021) have expanded all these previous observations to a diverse set of pathosystems that range from viruses, bacteria, oomycetes, fungi, nematodes to insects. While modularity was barely seen in only a few of the systems, nestedness was a common trend (observed in ~94% of all systems). This nestedness, as seen in previous studies and as predicted by theory, emerged as a consequence of the existence of generalist and specialist strains of the parasites that differed in their capacity to infect more or less resistant plant genotypes. As pointed out by Moury et al. (2021) in their conclusions, the ubiquity of nestedness in plant-parasite infection matrices has strong implications for the evolution and management of infectious diseases. References Flor, H. H. (1956). The complementary genic systems in flax and flax rust. In Advances in genetics, 8, 29-54. https://doi.org/10.1016/S0065-2660(08)60498-8 Flores, C. O., Meyer, J. R., Valverde, S., Farr, L., and Weitz, J. S. (2011). Statistical structure of host–phage interactions. Proceedings of the National Academy of Sciences, 108, E288-E297. https://doi.org/10.1073/pnas.1101595108 Frank, S. A. (2000). Specific and non-specific defense against parasitic attack. Journal of Theoretical Biology, 202, 283-304. https://doi.org/10.1006/jtbi.1999.1054 González, R., Butković, A., and Elena, S. F. (2019). Role of host genetic diversity for susceptibility-to-infection in the evolution of virulence of a plant virus. Virus evolution, 5(2), vez024. https://doi.org/10.1093/ve/vez052 Hillung, J., Cuevas, J. M., Valverde, S., and Elena, S. F. (2014). Experimental evolution of an emerging plant virus in host genotypes that differ in their susceptibility to infection. Evolution, 68, 2467-2480. https://doi.org/10.1111/evo.12458 McLeish, M., Sacristán, S., Fraile, A., and García-Arenal, F. (2019). Coinfection organizes epidemiological networks of viruses and hosts and reveals hubs of transmission. Phytopathology, 109, 1003-1010. https://doi.org/10.1094/PHYTO-08-18-0293-R Moury B, Audergon J-M, Baudracco-Arnas S, Krima SB, Bertrand F, Boissot N, Buisson M, Caffier V, Cantet M, Chanéac S, Constant C, Delmotte F, Dogimont C, Doumayrou J, Fabre F, Fournet S, Grimault V, Jaunet T, Justafré I, Lefebvre V, Losdat D, Marcel TC, Montarry J, Morris CE, Omrani M, Paineau M, Perrot S, Pilet-Nayel M-L and Ruellan Y (2021) The quasi-universality of nestedness in the structure of quantitative plant-parasite interactions. bioRxiv, 2021.03.03.433745, ver. 4 recommended and peer-reviewed by PCI Evolutionary Biology. https://doi.org/10.1101/2021.03.03.433745 Navarro, R., Ambros, S., Martinez, F., Wu, B., Carrasco, J. L., and Elena, S. F. (2020). Defects in plant immunity modulate the rates and patterns of RNA virus evolution. bioRxiv. doi: https://doi.org/10.1101/2020.10.13.337402 Valverde, S., Vidiella, B., Montañez, R., Fraile, A., Sacristán, S., and García-Arenal, F. (2020). Coexistence of nestedness and modularity in host–pathogen infection networks. Nature ecology & evolution, 4, 568-577. https://doi.org/10.1038/s41559-020-1130-9 | The quasi-universality of nestedness in the structure of quantitative plant-parasite interactions | Moury Benoît, Audergon Jean-Marc, Baudracco-Arnas Sylvie, Ben Krima Safa, Bertrand François, Boissot Nathalie, Buisson Mireille, Caffier Valérie, Cantet Mélissa, Chanéac Sylvia, Constant Carole, Delmotte François, Dogimont Catherine, Doumayrou Jul... | <p>Understanding the relationships between host range and pathogenicity for parasites, and between the efficiency and scope of immunity for hosts are essential to implement efficient disease control strategies. In the case of plant parasites, most... | | Bioinformatics & Computational Biology, Evolutionary Dynamics, Species interactions | Santiago Elena | 2021-03-04 21:23:08 | ||
20 Dec 2017

Renewed diversification following Miocene landscape turnover in a Neotropical butterfly radiationThe influence of environmental change over geological time on the tempo and mode of biological diversification, revealed by Neotropical butterfliesRecommended by Richard H Ree based on reviews by Delano Lewis and 1 anonymous reviewerThe influence of environmental change over geological time on the tempo and mode of biological diversification is a hot topic in biogeography. Of central interest are questions about where, when, and how fast lineages proliferated, suffered extinction, and migrated in response to tectonic events, the waxing and waning of dominant biomes, etc. In this context, the dynamic conditions of the Miocene have received much attention, from studies of many clades and biogeographic regions. Here, Chazot et al. [1] present an exemplary analysis of butterflies (tribe Ithomiini) in the Neotropics, examining their diversification across the Andes and Amazon. They infer sharp contrasts between these regions in the late Miocene: accelerated diversification during orogeny of the Andes, and greater extinction in the Amazon associated during the Pebas system, with interchange and local diversification increasing following the Pebas during the Pliocene. References [1] Chazot N, Willmott KR, Lamas G, Freitas AVL, Piron-Prunier F, Arias CF, Mallet J, De-Silva DL and Elias M. 2017. Renewed diversification following Miocene landscape turnover in a Neotropical butterfly radiation. BioRxiv 148189, ver 4 of 19th December 2017. doi: 10.1101/148189 [2] Xing Y, and Ree RH. 2017. Uplift-driven diversification in the Hengduan Mountains, a temperate biodiversity hotspot. Proceedings of the National Academy of Sciences of the United States of America, 114: E3444-E3451. doi: 10.1073/pnas.1616063114 | Renewed diversification following Miocene landscape turnover in a Neotropical butterfly radiation | Nicolas Chazot, Keith R. Willmott, Gerardo Lamas, André V.L. Freitas, Florence Piron-Prunier, Carlos F. Arias, James Mallet, Donna Lisa De-Silva, Marianne Elias | The Neotropical region has experienced a dynamic landscape evolution throughout the Miocene, with the large wetland Pebas occupying western Amazonia until 11-8 my ago and continuous uplift of the Andes mountains along the western edge of South Ame... | | Macroevolution, Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Richard H Ree | 2017-06-12 11:55:14 | ||
20 May 2020
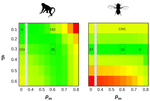
How much does Ne vary among species?Further questions on the meaning of effective population sizeRecommended by Martin Lascoux based on reviews by 3 anonymous reviewersIn spite of its name, the effective population size, Ne, has a complex and often distant relationship to census population size, as we usually understand it. In truth, it is primarily an abstract concept aimed at measuring the amount of genetic drift occurring in a population at any given time. The standard way to model random genetic drift in population genetics is the Wright-Fisher model and, with a few exceptions, definitions of the effective population size stems from it: “a certain model has effective population size, Ne, if some characteristic of the model has the same value as the corresponding characteristic for the simple Wright-Fisher model whose actual size is Ne” (Ewens 2004). Since Sewall Wright introduced the concept of effective population size in 1931 (Wright 1931), it has flourished and there are today numerous definitions of it depending on the process being examined (genetic diversity, loss of alleles, efficacy of selection) and the characteristic of the model that is considered. These different definitions of the effective population size were generally introduced to address specific aspects of the evolutionary process. One aspect that has been hotly debated since the first estimates of genetic diversity in natural populations were published is the so-called Lewontin’s paradox (1974). Lewontin noted that the observed variation in heterozygosity across species was much smaller than one would expect from the neutral expectations calculated with the actual size of the species. References Brandvain Y, Wright SI (2016) The Limits of Natural Selection in a Nonequilibrium World. Trends in Genetics, 32, 201–210. doi: 10.1016/j.tig.2016.01.004 | How much does Ne vary among species? | Nicolas Galtier, Marjolaine Rousselle | <p>Genetic drift is an important evolutionary force of strength inversely proportional to *Ne*, the effective population size. The impact of drift on genome diversity and evolution is known to vary among species, but quantifying this effect is a d... | | Bioinformatics & Computational Biology, Genome Evolution, Molecular Evolution, Population Genetics / Genomics | Martin Lascoux | 2019-12-08 00:11:00 | ||
03 Aug 2017
POSTPRINT
Fisher's geometrical model and the mutational patterns of antibiotic resistance across dose gradientsWhat doesn’t kill us makes us stronger: can Fisher’s Geometric model predict antibiotic resistance evolution?Recommended by Inês Fragata and Claudia BankThe increasing number of reported cases of antibiotic resistance is one of today’s major public health concerns. Dealing with this threat involves understanding what drives the evolution of antibiotic resistance and investigating whether we can predict (and subsequently avoid or circumvent) it [1]. References [1] Palmer AC, and Kishony R. 2013. Understanding, predicting and manipulating the genotypic evolution of antibiotic resistance. Nature Review Genetics 14: 243—248. doi: 10.1038/nrg3351 [2] Tenaillon O. 2014. The utility of Fisher’s geometric model in evolutionary genetics. Annual Review of Ecology, Evolution and Systematics 45: 179—201. doi: 10.1146/annurev-ecolsys-120213-091846 [3] Blanquart F and Bataillon T. 2016. Epistasis and the structure of fitness landscapes: are experimental fitness landscapes compatible with Fisher’s geometric model? Genetics 203: 847—862. doi: 10.1534/genetics.115.182691 [4] Harmand N, Gallet R, Jabbour-Zahab R, Martin G and Lenormand T. 2017. Fisher’s geometrical model and the mutational patterns of antibiotic resistance across dose gradients. Evolution 71: 23—37. doi: 10.1111/evo.13111 [5] de Visser, JAGM, and Krug J. 2014. Empirical fitness landscapes and the predictability of evolution. Nature 15: 480—490. doi: 10.1038/nrg3744 [6] Palmer AC, Toprak E, Baym M, Kim S, Veres A, Bershtein S and Kishony R. 2015. Delayed commitment to evolutionary fate in antibiotic resistance fitness landscapes. Nature Communications 6: 1—8. doi: 10.1038/ncomms8385 | Fisher's geometrical model and the mutational patterns of antibiotic resistance across dose gradients | Noémie Harmand, Romain Gallet, Roula Jabbour-Zahab, Guillaume Martin, Thomas Lenormand | Fisher's geometrical model (FGM) has been widely used to depict the fitness effects of mutations. It is a general model with few underlying assumptions that gives a large and comprehensive view of adaptive processes. It is thus attractive in sever... | | Adaptation | Inês Fragata | 2017-08-01 16:06:02 |
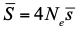 , where Ne is the effective population size and
, where Ne is the effective population size and  is the mean fitness effect of non-synonymous mutations. Assuming further that distinct species share a common DFE and therefore a common
is the mean fitness effect of non-synonymous mutations. Assuming further that distinct species share a common DFE and therefore a common  . Applying their newly developed approach to various datasets they conclude that the power of drift varies by a factor of at least 500 between large-Ne (Drosophila) and small-Ne species (H. sapiens). This is an order of magnitude larger than what would be obtained by comparing estimates of the variation in neutral diversity. Hence the proposed approach seems to have gone some way in making Lewontin’s paradox less paradoxical. But, perhaps more importantly, as the authors tersely point out at the end of the abstract their results further questions the meaning of Ne parameters in population genetics. And arguably this could well be the most important contribution of their study and something that is badly needed.
. Applying their newly developed approach to various datasets they conclude that the power of drift varies by a factor of at least 500 between large-Ne (Drosophila) and small-Ne species (H. sapiens). This is an order of magnitude larger than what would be obtained by comparing estimates of the variation in neutral diversity. Hence the proposed approach seems to have gone some way in making Lewontin’s paradox less paradoxical. But, perhaps more importantly, as the authors tersely point out at the end of the abstract their results further questions the meaning of Ne parameters in population genetics. And arguably this could well be the most important contribution of their study and something that is badly needed. 



