
Latest recommendations

| Id | Title | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers▲ | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
06 Apr 2021
How robust are cross-population signatures of polygenic adaptation in humans?Be careful when studying selection based on polygenic score overdispersionRecommended by Torsten Günther based on reviews by Lawrence Uricchio, Mashaal Sohail, Barbara Bitarello and 1 anonymous reviewer based on reviews by Lawrence Uricchio, Mashaal Sohail, Barbara Bitarello and 1 anonymous reviewer
The advent of genome-wide association studies (GWAS) has been a great promise for our understanding of the connection between genotype and phenotype. Today, the NHGRI-EBI GWAS catalog contains 251,401 associations from 4,961 studies (1). This wealth of studies has also generated interest to use the summary statistics beyond the few top hits in order to make predictions for individuals without known phenotype, e.g. to predict polygenic risk scores or to study polygenic selection by comparing different groups. For instance, polygenic selection acting on the most studied polygenic trait, height, has been subject to multiple studies during the past decade (e.g. 2–6). They detected north-south gradients in Europe which were consistent with expectations. However, their GWAS summary statistics were based on the GIANT consortium data set, a meta-analysis of GWAS conducted in different European cohorts (7,8). The availability of large data sets with less stratification such as the UK Biobank (9) has led to a re-evaluation of those results. The nature of the GIANT consortium data set was realized to represent a potential problem for studies of polygenic adaptation which led several of the authors of the original articles to caution against the interpretations of polygenic selection on height (10,11). This was a great example on how the scientific community assessed their own earlier results in a critical way as more data became available. At the same time it left the question whether there is detectable polygenic selection separating populations more open than ever. Generally, recent years have seen several articles critically assessing the portability of GWAS results and risk score predictions to other populations (12–14). Refoyo-Martínez et al. (15) are now presenting a systematic assessment on the robustness of cross-population signatures of polygenic adaptation in humans. They compiled GWAS results for complex traits which have been studied in more than one cohort and then use allele frequencies from the 1000 Genomes Project data (16) set to detect signals of polygenic score overdispersion. As the source for the allele frequencies is kept the same across all tests, differences between the signals must be caused by the underlying GWAS. The results are concerning as the level of overdispersion largely depends on the choice of GWAS cohort. Cohorts with homogenous ancestries show little to no overdispersion compared to cohorts of mixed ancestries such as meta-analyses. It appears that the meta-analyses fail to fully account for stratification in their data sets. The authors based most of their analyses on the heavily studied trait height. Additionally, they use educational attainment (measured as the number of school years of an individual) as an example. This choice was due to the potential over- or misinterpretation of results by the media, the general public and by far right hate groups. Such traits are potentially confounded by unaccounted cultural and socio-economic factors. Showing that previous results about polygenic selection on educational attainment are not robust is an important result that needs to be communicated well. This forms a great example for everyone working in human genomics. We need to be aware that our results can sometimes be misinterpreted. And we need to make an effort to write our papers and communicate our results in a way that is honest about the limitations of our research and that prevents the misuse of our results by hate groups. This article represents an important contribution to the field. It is cruicial to be aware of potential methodological biases and technical artifacts. Future studies of polygenic adaptation need to be cautious with their interpretations of polygenic score overdispersion. A recommendation would be to use GWAS results obtained in homogenous cohorts. But even if different biobank-scale cohorts of homogeneous ancestry are employed, there will always be some remaining risk of unaccounted stratification. These conclusions may seem sobering but they are part of the scientific process. We need additional controls and new, different methods than polygenic score overdispersion for assessing polygenic selection. Last year also saw the presentation of a novel approach using sequence data and GWAS summary statistics to detect directional selection on a polygenic trait (17). This new method appears to be robust to bias stemming from stratification in the GWAS cohort as well as other confounding factors. Such new developments show light at the end of the tunnel for the use of GWAS summary statistics in the study of polygenic adaptation. References 1. Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Research. 2019 Jan 8;47(D1):D1005–12. doi: https://doi.org/10.1093/nar/gky1120 2. Turchin MC, Chiang CW, Palmer CD, Sankararaman S, Reich D, Hirschhorn JN. Evidence of widespread selection on standing variation in Europe at height-associated SNPs. Nature Genetics. 2012 Sep;44(9):1015–9. doi: https://doi.org/10.1038/ng.2368 3. Berg JJ, Coop G. A Population Genetic Signal of Polygenic Adaptation. PLOS Genetics. 2014 Aug 7;10(8):e1004412. doi: https://doi.org/10.1371/journal.pgen.1004412 4. Robinson MR, Hemani G, Medina-Gomez C, Mezzavilla M, Esko T, Shakhbazov K, et al. Population genetic differentiation of height and body mass index across Europe. Nature Genetics. 2015 Nov;47(11):1357–62. doi: https://doi.org/10.1038/ng.3401 5. Mathieson I, Lazaridis I, Rohland N, Mallick S, Patterson N, Roodenberg SA, et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015 Dec;528(7583):499–503. doi: https://doi.org/10.1038/nature16152 6. Racimo F, Berg JJ, Pickrell JK. Detecting polygenic adaptation in admixture graphs. Genetics. 2018. Arp;208(4):1565–1584. doi: https://doi.org/10.1534/genetics.117.300489 7. Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010 Oct;467(7317):832–8. doi: https://doi.org/10.1038/nature09410 8. Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014 Nov;46(11):1173–86. doi: https://doi.org/10.1038/ng.3097 9. Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018 Oct;562(7726):203–9. doi: https://doi.org/10.1038/s41586-018-0579-z 10. Berg JJ, Harpak A, Sinnott-Armstrong N, Joergensen AM, Mostafavi H, Field Y, et al. Reduced signal for polygenic adaptation of height in UK Biobank. eLife. 2019 Mar 21;8:e39725. doi: https://doi.org/10.7554/eLife.39725 11. Sohail M, Maier RM, Ganna A, Bloemendal A, Martin AR, Turchin MC, et al. Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies. eLife. 2019 Mar 21;8:e39702. doi: https://doi.org/10.7554/eLife.39702 12. Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nature Genetics. 2019 Apr;51(4):584–91. doi: https://doi.org/10.1038/s41588-019-0379-x 13. Bitarello BD, Mathieson I. Polygenic Scores for Height in Admixed Populations. G3: Genes, Genomes, Genetics. 2020 Nov 1;10(11):4027–36. doi: https://doi.org/10.1534/g3.120.401658 14. Uricchio LH, Kitano HC, Gusev A, Zaitlen NA. An evolutionary compass for detecting signals of polygenic selection and mutational bias. Evolution Letters. 2019;3(1):69–79. doi: https://doi.org/10.1002/evl3.97 15. Refoyo-Martínez A, Liu S, Jørgensen AM, Jin X, Albrechtsen A, Martin AR, Racimo F. How robust are cross-population signatures of polygenic adaptation in humans? bioRxiv, 2021, 2020.07.13.200030, version 5 peer-reviewed and recommended by Peer community in Evolutionary Biology. doi: https://doi.org/10.1101/2020.07.13.200030 16. Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, et al. A global reference for human genetic variation. Nature. 2015 Sep 30;526(7571):68–74. doi: https://doi.org/10.1038/nature15393 17. Stern AJ, Speidel L, Zaitlen NA, Nielsen R. Disentangling selection on genetically correlated polygenic traits using whole-genome genealogies. bioRxiv. 2020 May 8;2020.05.07.083402. doi: https://doi.org/10.1101/2020.05.07.083402 | How robust are cross-population signatures of polygenic adaptation in humans? | Alba Refoyo-Martínez, Siyang Liu, Anja Moltke Jørgensen, Xin Jin, Anders Albrechtsen, Alicia R. Martin, Fernando Racimo | <p>Over the past decade, summary statistics from genome-wide association studies (GWASs) have been used to detect and quantify polygenic adaptation in humans. Several studies have reported signatures of natural selection at sets of SNPs associated... | | Bioinformatics & Computational Biology, Genetic conflicts, Human Evolution, Population Genetics / Genomics | Torsten Günther | 2020-08-14 15:06:54 | ||
23 Jun 2021

Evolution of flowering time in a selfing annual plant: Roles of adaptation and genetic driftSeparating adaptation from drift: A cautionary tale from a self-fertilizing plantRecommended by Christoph Haag based on reviews by Pierre Olivier Cheptou, Jon Agren and Stefan LaurentIn recent years many studies have documented shifts in phenology in response to climate change, be it in arrival times in migrating birds, budset in trees, adult emergence in butterflies, or flowering time in annual plants (Coen et al. 2018; Piao et al. 2019). While these changes are, in part, explained by phenotypic plasticity, more and more studies find that they involve also genetic changes, that is, they involve evolutionary change (e.g., Metz et al. 2020). Yet, evolutionary change may occur through genetic drift as well as selection. Therefore, in order to demonstrate adaptive evolutionary change in response to climate change, drift has to be excluded as an alternative explanation (Hansen et al. 2012). A new study by Gay et al. (2021) shows just how difficult this can be. The authors investigated a recent evolutionary shift in flowering time by in a population an annual plant that reproduces predominantly by self-fertilization. The population has recently been subjected to increased temperatures and reduced rainfalls both of which are believed to select for earlier flowering times. They used a “resurrection” approach (Orsini et al. 2013; Weider et al. 2018): Genotypes from the past (resurrected from seeds) were compared alongside more recent genotypes (from more recently collected seeds) under identical conditions in the greenhouse. Using an experimental design that replicated genotypes, eliminated maternal effects, and controlled for microenvironmental variation, they found said genetic change in flowering times: Genotypes obtained from recently collected seeds flowered significantly (about 2 days) earlier than those obtained 22 generations before. However, neutral markers (microsatellites) also showed strong changes in allele frequencies across the 22 generations, suggesting that effective population size, Ne, was low (i.e., genetic drift was strong), which is typical for highly self-fertilizing populations. In addition, several multilocus genotypes were present at high frequencies and persisted over the 22 generations, almost as in clonal populations (e.g., Schaffner et al. 2019). The challenge was thus to evaluate whether the observed evolutionary change was the result of an adaptive response to selection or may be explained by drift alone. Here, Gay et al. (2021) took a particularly careful and thorough approach. First, they carried out a selection gradient analysis, finding that earlier-flowering plants produced more seeds than later-flowering plants. This suggests that, under greenhouse conditions, there was indeed selection for earlier flowering times. Second, investigating other populations from the same region (all populations are located on the Mediterranean island of Corsica, France), they found that a concurrent shift to earlier flowering times occurred also in these populations. Under the hypothesis that the populations can be regarded as independent replicates of the evolutionary process, the observation of concurrent shifts rules out genetic drift (under drift, the direction of change is expected to be random). The study may well have stopped here, concluding that there is good evidence for an adaptive response to selection for earlier flowering times in these self-fertilizing plants, at least under the hypothesis that selection gradients estimated in the greenhouse are relevant to field conditions. However, the authors went one step further. They used the change in the frequencies of the multilocus genotypes across the 22 generations as an estimate of realized fitness in the field and compared them to the phenotypic assays from the greenhouse. The results showed a tendency for high-fitness genotypes (positive frequency changes) to flower earlier and to produce more seeds than low-fitness genotypes. However, a simulation model showed that the observed correlations could be explained by drift alone, as long as Ne is lower than ca. 150 individuals. The findings were thus consistent with an adaptive evolutionary change in response to selection, but drift could only be excluded as the sole explanation if the effective population size was large enough. The study did provide two estimates of Ne (19 and 136 individuals, based on individual microsatellite loci or multilocus genotypes, respectively), but both are problematic. First, frequency changes over time may be influenced by the presence of a seed bank or by immigration from a genetically dissimilar population, which may lead to an underestimation of Ne (Wang and Whitlock 2003). Indeed, the low effective size inferred from the allele frequency changes at microsatellite loci appears to be inconsistent with levels of genetic diversity found in the population. Moreover, high self-fertilization reduces effective recombination and therefore leads to non-independence among loci. This lowers the precision of the Ne estimates (due to a higher sampling variance) and may also violate the assumption of neutrality due to the possibility of selection (e.g., due to inbreeding depression) at linked loci, which may be anywhere in the genome in case of high degrees of self-fertilization. There is thus no definite answer to the question of whether or not the observed changes in flowering time in this population were driven by selection. The study sets high standards for other, similar ones, in terms of thoroughness of the analyses and care in interpreting the findings. It also serves as a very instructive reminder to carefully check the assumptions when estimating neutral expectations, especially when working on species with complicated demographies or non-standard life cycles. Indeed the issues encountered here, in particular the difficulty of establishing neutral expectations in species with low effective recombination, may apply to many other species, including partially or fully asexual ones (Hartfield 2016). Furthermore, they may not be limited to estimating Ne but may also apply, for instance, to the establishment of neutral baselines for outlier analyses in genome scans (see e.g, Orsini et al. 2012). References Cohen JM, Lajeunesse MJ, Rohr JR (2018) A global synthesis of animal phenological responses to climate change. Nature Climate Change, 8, 224–228. https://doi.org/10.1038/s41558-018-0067-3 Gay L, Dhinaut J, Jullien M, Vitalis R, Navascués M, Ranwez V, Ronfort J (2021) Evolution of flowering time in a selfing annual plant: Roles of adaptation and genetic drift. bioRxiv, 2020.08.21.261230, ver. 4 recommended and peer-reviewed by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.08.21.261230 Hansen MM, Olivieri I, Waller DM, Nielsen EE (2012) Monitoring adaptive genetic responses to environmental change. Molecular Ecology, 21, 1311–1329. https://doi.org/10.1111/j.1365-294X.2011.05463.x Hartfield M (2016) Evolutionary genetic consequences of facultative sex and outcrossing. Journal of Evolutionary Biology, 29, 5–22. https://doi.org/10.1111/jeb.12770 Metz J, Lampei C, Bäumler L, Bocherens H, Dittberner H, Henneberg L, Meaux J de, Tielbörger K (2020) Rapid adaptive evolution to drought in a subset of plant traits in a large-scale climate change experiment. Ecology Letters, 23, 1643–1653. https://doi.org/10.1111/ele.13596 Orsini L, Schwenk K, De Meester L, Colbourne JK, Pfrender ME, Weider LJ (2013) The evolutionary time machine: using dormant propagules to forecast how populations can adapt to changing environments. Trends in Ecology & Evolution, 28, 274–282. https://doi.org/10.1016/j.tree.2013.01.009 Orsini L, Spanier KI, Meester LD (2012) Genomic signature of natural and anthropogenic stress in wild populations of the waterflea Daphnia magna: validation in space, time and experimental evolution. Molecular Ecology, 21, 2160–2175. https://doi.org/10.1111/j.1365-294X.2011.05429.x Piao S, Liu Q, Chen A, Janssens IA, Fu Y, Dai J, Liu L, Lian X, Shen M, Zhu X (2019) Plant phenology and global climate change: Current progresses and challenges. Global Change Biology, 25, 1922–1940. https://doi.org/10.1111/gcb.14619 Schaffner LR, Govaert L, De Meester L, Ellner SP, Fairchild E, Miner BE, Rudstam LG, Spaak P, Hairston NG (2019) Consumer-resource dynamics is an eco-evolutionary process in a natural plankton community. Nature Ecology & Evolution, 3, 1351–1358. https://doi.org/10.1038/s41559-019-0960-9 Wang J, Whitlock MC (2003) Estimating Effective Population Size and Migration Rates From Genetic Samples Over Space and Time. Genetics, 163, 429–446. PMID: 12586728 Weider LJ, Jeyasingh PD, Frisch D (2018) Evolutionary aspects of resurrection ecology: Progress, scope, and applications—An overview. Evolutionary Applications, 11, 3–10. https://doi.org/10.1111/eva.12563 | Evolution of flowering time in a selfing annual plant: Roles of adaptation and genetic drift | Laurène Gay, Julien Dhinaut, Margaux Jullien, Renaud Vitalis, Miguel Navascués, Vincent Ranwez, and Joëlle Ronfort | <p style="text-align: justify;">Resurrection studies are a useful tool to measure how phenotypic traits have changed in populations through time. If these traits modifications correlate with the environmental changes that occurred during the time ... | | Adaptation, Evolutionary Ecology, Genotype-Phenotype, Phenotypic Plasticity, Population Genetics / Genomics, Quantitative Genetics, Reproduction and Sex | Christoph Haag | 2020-08-21 17:26:59 | ||
01 Mar 2021
Social Conflicts in Dictyostelium discoideum : A Matter of ScalesThe cell-level perspective in social conflicts in Dictyostelium discoideumRecommended by Jeremy Van Cleve based on reviews by Peter Conlin and ?The social amoeba Dictyostelium discoideum is an important model system for the study of cooperation and multicellularity as is has both unicellular and aggregative life phases. In the aggregative phase, which typically occurs when nutrients are limiting, individual cells eventually gather together to form a fruiting bodies whose spores may be dispersed to another, better, location and whose stalk cells, which support the spores, die. This extreme form of cooperation has been the focus of numerous studies that have revealed the importance genetic relatedness and kin selection (Hamilton 1964; Lehmann and Rousset 2014) in explaining the maintenance of this cooperative collective behavior (Strassmann et al. 2000; Kuzdzal-Fick et al. 2011; Strassmann and Queller 2011). However, much remains unknown with respect to how the interactions between individual cells, their neighbors, and their environment produce cooperative behavior at the scale of whole groups or collectives. In this preprint, Forget et al. (2021) describe how the D. discoideum system is crucial in this respect because it allows these cellular-level interactions to be studied in a systematic and tractable manner. References Forget, M., Adiba, S. and De Monte, S.(2021) Social conflicts in *Dictyostelium discoideum *: a matter of scales. HAL, hal-03088868, ver. 2 peer-reviewed and recommended by PCI Evolutionary Biology. https://hal.archives-ouvertes.fr/hal-03088868/ Hamilton, W. D. (1964). The genetical evolution of social behaviour. II. Journal of theoretical biology, 7(1), 17-52. doi: https://doi.org/10.1016/0022-5193(64)90039-6 Kuzdzal-Fick, J. J., Fox, S. A., Strassmann, J. E., and Queller, D. C. (2011). High relatedness is necessary and sufficient to maintain multicellularity in Dictyostelium. Science, 334(6062), 1548-1551. doi: https://doi.org/10.1126/science.1213272 Lehmann, L., and Rousset, F. (2014). The genetical theory of social behaviour. Philosophical Transactions of the Royal Society B: Biological Sciences, 369(1642), 20130357. doi: https://doi.org/10.1098/rstb.2013.0357 Strassmann, J. E., and Queller, D. C. (2011). Evolution of cooperation and control of cheating in a social microbe. Proceedings of the National Academy of Sciences, 108(Supplement 2), 10855-10862. doi: https://doi.org/10.1073/pnas.1102451108 Strassmann, J. E., Zhu, Y., & Queller, D. C. (2000). Altruism and social cheating in the social amoeba Dictyostelium discoideum. Nature, 408(6815), 965-967. doi: https://doi.org/10.1038/35050087 Thompson, C. R., & Kay, R. R. (2000). Cell-fate choice in Dictyostelium: intrinsic biases modulate sensitivity to DIF signaling. Developmental biology, 227(1), 56-64. doi: https://doi.org/10.1006/dbio.2000.9877 | Social Conflicts in Dictyostelium discoideum : A Matter of Scales | Forget, Mathieu; Adiba, Sandrine; De Monte, Silvia | <p>The 'social amoeba' Dictyostelium discoideum, where aggregation of genetically heterogeneous cells produces functional collective structures, epitomizes social conflicts associated with multicellular organization. 'Cheater' populations that hav... | | Behavior & Social Evolution, Evolutionary Dynamics, Evolutionary Theory, Experimental Evolution | Jeremy Van Cleve | 2020-08-28 10:37:21 | ||
04 Mar 2021
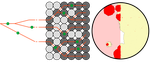
Simulation of bacterial populations with SLiMSimulating bacterial evolution forward-in-timeRecommended by Frederic Bertels based on reviews by 3 anonymous reviewersJean Cury and colleagues (2021) have developed a protocol to simulate bacterial evolution in SLiM. In contrast to existing methods that depend on the coalescent, SLiM simulates evolution forward in time. SLiM has, up to now, mostly been used to simulate the evolution of eukaryotes (Haller and Messer 2019), but has been adapted here to simulate evolution in bacteria. Forward-in-time simulations are usually computationally very costly. To circumvent this issue, bacterial population sizes are scaled down. One would now expect results to become inaccurate, however, Cury et al. show that scaled-down forwards simulations provide very accurate results (similar to those provided by coalescent simulators) that are consistent with theoretical expectations. Simulations were analyzed and compared to existing methods in simple and slightly more complex scenarios where recombination affects evolution. In all scenarios, simulation results from coalescent methods (fastSimBac (De Maio and Wilson 2017), ms (Hudson 2002)) and scaled-down forwards simulations were very similar, which is very good news indeed. A biologist not aware of the complexities of forwards, backwards simulations and the coalescent, might now naïvely ask why another simulation method is needed if existing methods perform just as well. To address this question the manuscript closes with a very neat example of what exactly is possible with forwards simulations that cannot be achieved using existing methods. The situation modeled is the growth and evolution of a set of 50 bacteria that are randomly distributed on a petri dish. One side of the petri dish is covered in an antibiotic the other is antibiotic-free. Over time, the bacteria grow and acquire antibiotic resistance mutations until the entire artificial petri dish is covered with a bacterial lawn. This simulation demonstrates that it is possible to simulate extremely complex (e.g. real world) scenarios to, for example, assess whether certain phenomena are expected with our current understanding of bacterial evolution, or whether there are additional forces that need to be taken into account. Hence, forwards simulators could significantly help us to understand what current models can and cannot explain in evolutionary biology.
References Cury J, Haller BC, Achaz G, Jay F (2021) Simulation of bacterial populations with SLiM. bioRxiv, 2020.09.28.316869, version 5 peer-reviewed and recommended by Peer community in Evolutionary Biology. https://doi.org/10.1101/2020.09.28.316869 De Maio N, Wilson DJ (2017) The Bacterial Sequential Markov Coalescent. Genetics, 206, 333–343. https://doi.org/10.1534/genetics.116.198796 Haller BC, Messer PW (2019) SLiM 3: Forward Genetic Simulations Beyond the Wright–Fisher Model. Molecular Biology and Evolution, 36, 632–637. https://doi.org/10.1093/molbev/msy228 Hudson RR (2002) Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics, 18, 337–338. https://doi.org/10.1093/bioinformatics/18.2.337 | Simulation of bacterial populations with SLiM | Jean Cury, Benjamin C. Haller, Guillaume Achaz, and Flora Jay | <p>Simulation of genomic data is a key tool in population genetics, yet, to date, there is no forward-in-time simulator of bacterial populations that is both computationally efficient and adaptable to a wide range of scenarios. Here we demonstrate... | | Bioinformatics & Computational Biology, Population Genetics / Genomics | Frederic Bertels | 2020-10-02 19:03:42 | ||
25 Feb 2021

Alteration of gut microbiota with a broad-spectrum antibiotic does not impair maternal care in the European earwigAssessing the role of host-symbiont interactions in maternal care behaviourRecommended by Trine Bilde based on reviews by Nadia Aubin-Horth, Gabrielle Davidson and 1 anonymous reviewer
The role of microbial symbionts in governing social traits of their hosts is an exciting and developing research area. Just like symbionts influence host reproductive behaviour and can cause mating incompatibilities to promote symbiont transmission through host populations (Engelstadter and Hurst 2009; Correa and Ballard 2016; Johnson and Foster 2018) (see also discussion on conflict resolution in Johnsen and Foster 2018), microbial symbionts could enhance transmission by promoting the social behaviour of their hosts (Ezenwa et al. 2012; Lewin-Epstein et al. 2017; Gurevich et al. 2020). Here I apply the term ‘symbiosis’ in the broad sense, following De Bary 1879 as “the living together of two differently named organisms“ independent of effects on the organisms involved (De Bary 1879), i.e. the biological interaction between the host and its symbionts may include mutualism, parasitism and commensalism. References Correa, C. C., and Ballard, J. W. O. (2016). Wolbachia associations with insects: winning or losing against a master manipulator. Frontiers in Ecology and Evolution, 3, 153. doi: https://doi.org/10.3389/fevo.2015.00153 De Bary, A. (1879). Die Erscheinung der Symbiose. Verlag von Karl J. Trubner, Strassburg. Engelstädter, J., and Hurst, G. D. (2009). The ecology and evolution of microbes that manipulate host reproduction. Annual Review of Ecology, Evolution, and Systematics, 40, 127-149. doi: https://doi.org/10.1146/annurev.ecolsys.110308.120206 Ezenwa, V. O., Gerardo, N. M., Inouye, D. W., Medina, M., and Xavier, J. B. (2012). Animal behavior and the microbiome. Science, 338(6104), 198-199. doi: https://doi.org/10.1126/science.1227412 Gurevich, Y., Lewin-Epstein, O., and Hadany, L. (2020). The evolution of paternal care: a role for microbes?. Philosophical Transactions of the Royal Society B, 375(1808), 20190599. doi: https://doi.org/10.1098/rstb.2019.0599 Hird, S. M. (2017). Evolutionary biology needs wild microbiomes. Frontiers in microbiology, 8, 725. doi: https://doi.org/10.3389/fmicb.2017.00725 Johnson, K. V. A., and Foster, K. R. (2018). Why does the microbiome affect behaviour?. Nature reviews microbiology, 16(10), 647-655. doi: https://doi.org/10.1038/s41579-018-0014-3 Kramer et al. (2017). When earwig mothers do not care to share: parent–offspring competition and the evolution of family life. Functional Ecology, 31(11), 2098-2107. doi: https://doi.org/10.1111/1365-2435.12915 Lewin-Epstein, O., Aharonov, R., and Hadany, L. (2017). Microbes can help explain the evolution of host altruism. Nature communications, 8(1), 1-7. doi: https://doi.org/10.1038/ncomms14040 Meunier, J., and Kölliker, M. (2012). Parental antagonism and parent–offspring co-adaptation interact to shape family life. Proceedings of the Royal Society B: Biological Sciences, 279(1744), 3981-3988. doi: https://doi.org/10.1098/rspb.2012.1416 Meunier, J., Wong, J. W., Gómez, Y., Kuttler, S., Röllin, L., Stucki, D., and Kölliker, M. (2012). One clutch or two clutches? Fitness correlates of coexisting alternative female life-histories in the European earwig. Evolutionary Ecology, 26(3), 669-682. doi: https://doi.org/10.1007/s10682-011-9510-x Nalepa, C. A. (2020). Origin of mutualism between termites and flagellated gut protists: transition from horizontal to vertical transmission. Frontiers in Ecology and Evolution, 8, 14. doi: https://doi.org/10.3389/fevo.2020.00014 Ratz, T., Kramer, J., Veuille, M., and Meunier, J. (2016). The population determines whether and how life-history traits vary between reproductive events in an insect with maternal care. Oecologia, 182(2), 443-452. doi: https://doi.org/10.1007/s00442-016-3685-3 Van Meyel, S., Devers, S., Dupont, S., Dedeine, F. and Meunier, J. (2021) Alteration of gut microbiota with a broad-spectrum antibiotic does not impair maternal care in the European earwig. bioRxiv, 2020.10.08.331363. ver. 5 peer-reviewed and recommended by PCI Evol Biol. https://doi.org/10.1101/2020.10.08.331363 | Alteration of gut microbiota with a broad-spectrum antibiotic does not impair maternal care in the European earwig | Sophie Van Meyel, Séverine Devers, Simon Dupont, Franck Dedeine and Joël Meunier | <p>The microbes residing within the gut of an animal host often increase their own fitness by modifying their host’s physiological, reproductive, and behavioural functions. Whereas recent studies suggest that they may also shape host sociality and... | | Behavior & Social Evolution, Evolutionary Ecology, Experimental Evolution, Life History, Species interactions | Trine Bilde | 2020-10-09 14:07:47 | ||
17 May 2021

Relative time constraints improve molecular datingDating with constraintsRecommended by Cécile Ané based on reviews by David Duchêne and 1 anonymous reviewerEstimating the absolute age of diversification events is challenging, because molecular sequences provide timing information in units of substitutions, not years. Additionally, the rate of molecular evolution (in substitutions per year) can vary widely across lineages. Accurate dating of speciation events traditionally relies on non-molecular data. For very fast-evolving organisms such as SARS-CoV-2, for which samples are obtained over a time span, the collection times provide this external information from which we can learn the rate of molecular evolution and date past events (Boni et al. 2020). In groups for which the fossil record is abundant, state-of-the-art dating methods use fossil information to complement molecular data, either in the form of a prior distribution on node ages (Nguyen & Ho 2020), or as data modelled with a fossilization process (Heath et al. 2014). Dating is a challenge in groups that lack fossils or other geological evidence, such as very old lineages and microbial lineages. In these groups, horizontal gene transfer (HGT) events have been identified as informative about relative dates: the ancestor of the gene's donor must be older than the descendants of the gene's recipient. Previous work using HGTs to date phylogenies have used methodologies that are ad-hoc (Davín et al 2018) or employ a small number of HGTs only (Magnabosco et al. 2018, Wolfe & Fournier 2018). Szöllősi et al. (2021) present and validate a Bayesian approach to estimate the age of diversification events based on relative information on these ages, such as implied by HGTs. This approach is flexible because it is modular: constraints on relative node ages can be combined with absolute age information from fossil data, and with any substitution model of molecular evolution, including complex state-of-art models. To ease the computational burden, the authors also introduce a two-step approach, in which the complexity of estimating branch lengths in substitutions per site is decoupled from the complexity of timing the tree with branch lengths in years, accounting for uncertainty in the first step. Currently, one limitation is that the tree topology needs to be known, and another limitation is that constraints need to be certain. Users of this method should be mindful of the latter when hundreds of constraints are used, as done by Szöllősi et al. (2021) to date the trees of Cyanobacteria and Archaea. Szöllősi et al. (2021)'s method is implemented in RevBayes, a highly modular platform for phylogenetic inference, rapidly growing in popularity (Höhna et al. 2016). The RevBayes tutorial page features a step-by-step tutorial "Dating with Relative Constraints", which makes the method highly approachable. References: Boni MF, Lemey P, Jiang X, Lam TT-Y, Perry BW, Castoe TA, Rambaut A, Robertson DL (2020) Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nature Microbiology, 5, 1408–1417. https://doi.org/10.1038/s41564-020-0771-4 Davín AA, Tannier E, Williams TA, Boussau B, Daubin V, Szöllősi GJ (2018) Gene transfers can date the tree of life. Nature Ecology & Evolution, 2, 904–909. https://doi.org/10.1038/s41559-018-0525-3 Heath TA, Huelsenbeck JP, Stadler T (2014) The fossilized birth–death process for coherent calibration of divergence-time estimates. Proceedings of the National Academy of Sciences, 111, E2957–E2966. https://doi.org/10.1073/pnas.1319091111 Höhna S, Landis MJ, Heath TA, Boussau B, Lartillot N, Moore BR, Huelsenbeck JP, Ronquist F (2016) RevBayes: Bayesian Phylogenetic Inference Using Graphical Models and an Interactive Model-Specification Language. Systematic Biology, 65, 726–736. https://doi.org/10.1093/sysbio/syw021 Magnabosco C, Moore KR, Wolfe JM, Fournier GP (2018) Dating phototrophic microbial lineages with reticulate gene histories. Geobiology, 16, 179–189. https://doi.org/10.1111/gbi.12273 Nguyen JMT, Ho SYW (2020) Calibrations from the Fossil Record. In: The Molecular Evolutionary Clock: Theory and Practice (ed Ho SYW), pp. 117–133. Springer International Publishing, Cham. https://doi.org/10.1007/978-3-030-60181-2_8 Szollosi, G.J., Hoehna, S., Williams, T.A., Schrempf, D., Daubin, V., Boussau, B. (2021) Relative time constraints improve molecular dating. bioRxiv, 2020.10.17.343889, ver. 8 recommended and peer-reviewed by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.10.17.343889 Wolfe JM, Fournier GP (2018) Horizontal gene transfer constrains the timing of methanogen evolution. Nature Ecology & Evolution, 2, 897–903. https://doi.org/10.1038/s41559-018-0513-7 | Relative time constraints improve molecular dating | Gergely J Szollosi, Sebastian Hoehna, Tom A Williams, Dominik Schrempf, Vincent Daubin, Bastien Boussau | <p style="text-align: justify;">Dating the tree of life is central to understanding the evolution of life on Earth. Molecular clocks calibrated with fossils represent the state of the art for inferring the ages of major groups. Yet, other informat... | | Bioinformatics & Computational Biology, Genome Evolution, Phylogenetics / Phylogenomics | Cécile Ané | 2020-10-21 23:39:17 | ||
04 Jul 2022
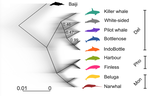
A genomic assessment of the marine-speciation paradox within the toothed whale superfamily DelphinoideaReticulated evolution marks the rapid diversification of the DelphinoidaeRecommended by Michael C. Fontaine based on reviews by Christelle Fraïsse, Simon Henry Martin, Andrew Foote and 2 anonymous reviewersHistorically neglected or considered a rare aberration in animals under the biological species concept, interspecific hybridisation has by now been recognised to be taxonomically widespread, particularly in rapidly diversifying groups (Dagilis et al. 2021; Edelman & Mallet 2021; Mallet et al. 2016; Seehausen 2004). Yet the prevalence of introgressive hybridizations, its evolutionary significance, and its impact on species diversification remain a hot topic of research in evolutionary biology. The rapid increase in genomic resources now available for non-model species has significantly contributed to the detection of introgressive hybridization across taxa showing that reticulated evolution is far more common in the animal kingdom than historically considered. Yet, detecting it, quantifying its magnitude, and assessing its evolutionary significance remains a challenging endeavour with constantly evolving methodologies to better explore and exploit genomic data (Blair & Ané 2020; Degnan & Rosenberg 2009; Edelman & Mallet 2021; Hibbins & Hahn 2022). In the marine realm, the dearth of geographic barriers and the large dispersal abilities of pelagic species like cetaceans have raised the questions of how populations and species can diverge and adapt to distinct ecological conditions in face of potentially large gene-flow, the so-called marine speciation paradox (Bierne et al. 2003). Contemporaneous hybridization among cetacean species has been widely documented in nature despite large phenotypic differences (Crossman et al. 2016). The historical prevalence of reticulated evolution, its evolutionary significance, and how it might have impacted the evolutionary history and diversification of the cetaceans have however remained elusive so far. Recent phylogenomic studies suggested that introgression has been prevalent in cetacean evolutionary history with instances reported among baleen whales (mysticetes) (Árnason et al. 2018) and among toothed whales (odontocetes), especially in the rapidly diversifying dolphins family of the Delphininae (Guo et al. 2021; Moura et al. 2020). Analysing publicly available whole-genome data from nine cetacean species across three families of Delphinoidae – dolphins, porpoises, and monondontidae – using phylogenomics and demo-genetics approaches, Westbury and colleagues (2022) take a step further in documenting that evolution among these species has been far from a simple bifurcating tree. Instead, their study describes widespread occurrences of introgression among Delphinoidae, drawing a complex picture of reticulated evolutionary history. After describing major topology discordance in phylogenetic gene trees along the genome, the authors use a panel of approaches to disentangle introgression from incomplete lineage sorting (ILS), the two most common causes of tree topology discordances (Hibbins & Hahn 2022). Applying popular tests that separate introgression from ILS, such as the Patterson’s D (a.k.a. ABBA-BABA) test (Durand et al. 2011; Green et al. 2010), QuIBL (Edelman et al. 2019), and D-FOIL (Pease & Hahn 2015), the authors report that signals of introgression are present in the genomes of most (if not all) the cetacean species included in their study. However, this picture needs to be nuanced. Most introgression signals seem to echo old introgression events that occurred primarily among ancestors. This is suggested by the differential signals of topology discordance along the genome when considering sliding windows along the genome of varying sizes (50kb, 100kb, and 1Mb), and by patterns of excess derived allele sharing along branches of the species tree, as captured by the f-branch test (Malinsky et al. 2021; Malinsky et al. 2018). The authors further investigated the dynamic of cessation of gene flow (and/or ILS) between species using the F1 hybrid PSMC (or hPSMC) approach (Cahill et al. 2016). By estimating the cross-coalescent rates (CRR) between species pairs with time in light of previously estimated species divergence times (McGowen et al. 2020), the authors report that gene flow (and/or ILS) most likely has stopped by now among most species, but it may have lasted for more than half of the time since the species split from each other. According to the author, this result may reflect the slow process by which reproductive isolation would have evolved between cetacean lineages, and that species isolation was marked by significant introgression events. Now, while the present study provides a good overview of how complex is the reticulated evolutionary history of the Delphinoidae, getting a complete picture will require overcoming a few important limitations. The first ones are methodological and related to the phylogenomic analyses. Given the sampling design with one diploid genome per species, the authors could not phase the data into the parental haplotypes, but instead relied on a majority consensus creating mosaic haploidized genomes representing a mixture between the two parental copies. Moreover, by using large genomic windows (≥50kb) that likely span multiple independent loci, phylogenetic analyses in windows encompassed distinct phylogenetic signals, potentially leading to bias and inaccuracy in the inferences. Thawornwattana et al (2018) previously showed that this “concatenation approach” could significantly impact phylogenetic inferences. They proposed instead to use loci small enough to minimise the risk of intra-locus recombination and to consider them in blocks of non-recombining loci along the genome in order to conduct phylogenetic analysed, ideally under the multi-species coalescent (MSC) that can account for ILS (e.g. BPP; Flouri et al. 2018; Jiao et al. 2020; Yang 2015). Such an approach applied to the diversification of the Delphinidae may reveal substantial changes compared to the currently admitted species tree. Inaccuracy in the species tree estimation may have a major impact on the introgression analyses conducted in this study since the species tree and branching order must be supplied in the introgression analyses to properly disentangle introgression from ILS. Here, the authors rely on the tree topology that was previously estimated in McGowen et al. (2020), which they also recovered using their consensus estimation from ASTRAL-III (Zhang et al. 2018). While the methodologies accounted to a certain extent for ILS, albeit with potential bias induced by the concatenation approach, they ignore the presumably large amount of introgression among species during the diversification process. Estimating species branching order while ignoring introgression can lead to major bias in the phylogenetic inference and can lead to incorrect topologies. Even if these MSC-based methods account for ILS, inferences can become very inaccurate or even break down as gene flow increases (see for ex. Jiao et al. 2020; Müller et al. in press; Solís-Lemus et al. 2016). Dedicated approaches have been developed to model explicitly introgression together with ILS to estimate phylogenetic networks (Blair & Ané 2020; Rabier et al. 2021) or in isolation-with-migration model (Müller et al. in press; Wang et al. 2020). Future studies revisiting the reticulated evolutionary history of the Delphinoidae with such approaches may not only precise the species branching order, but also deliver a finer view of the magnitude and prevalence of introgression during the evolutionary history of these species. A final part of Westbury et al. (2022)'s study set out to test whether historical periods of low abundance could have facilitated hybridization among Delphinoidae species. During these periods of low abundance, species may encounter only a limited number of conspecifics and may consider individuals from other species as suitable mating partners, leading to hybridisation (Crossman et al. 2016; Edwards et al. 2011; Westbury et al. 2019). The authors tested this hypothesis by considering genome-wide genetic diversity (or heterozygosity) as a proxy of historical effective population size (Ne), itself as a proxy of the evolution of census size with time. They also try to link historical Ne variation (from PSMC, Li & Durbin 2011) with their estimated time to cessation of gene flow or ILS (from the CRR of hPSMC). However, no straightforward relationship was found between the genetic diversity and the propensity of species to hybridize, nor was there any clear link between Ne variation through time and the cessation of gene flow or ILS. Such a lack of relationship may not come as a surprise, since the determinants of genome-wide genetic diversity and its variation through evolutionary time-scale are far more diverse and complex than just a direct link with hybridization, introgression, or even with the census population size. In fact, genetic diversity results from the balance between all the evolutionary processes at play in the species' evolutionary history (see the review of Ellegren & Galtier 2016). Other important factors can strongly impact genetic diversity, including demography and structure, but also linked selection (Boitard et al. 2022; Buffalo 2021; Ellegren & Galtier 2016). All in all, Westbury and coll. (2022) present here an interesting study providing an additional step towards resolving and understanding the complex evolutionary history of the Delphinoidae, and shedding light on the importance of introgression during the diversification of these cetacean species. Prospective work improving upon the taxonomic sampling, with additional genomic data for each species, considered with dedicated approaches tailored at estimating species tree or network while accounting for ILS and introgression will be key for refining the picture depicted in this study. Down the road, altogether these studies will contribute to assessing the evolutionary significance of introgression on the diversification of Delphinoides, and more generally on the diversification of cetacean species, which still remain an open and exciting perspective. References Árnason Ú, Lammers F, Kumar V, Nilsson MA, Janke A (2018) Whole-genome sequencing of the blue whale and other rorquals finds signatures for introgressive gene flow. Science Advances 4, eaap9873. https://doi.org/10.1126/sciadv.aap9873 Bierne N, Bonhomme F, David P (2003) Habitat preference and the marine-speciation paradox. Proceedings of the Royal Society of London. Series B: Biological Sciences 270, 1399-1406. https://doi.org/10.1098/rspb.2003.2404 Blair C, Ané C (2020) Phylogenetic Trees and Networks Can Serve as Powerful and Complementary Approaches for Analysis of Genomic Data. Systematic Biology 69, 593-601. https://doi.org/10.1093/sysbio/syz056 Boitard S, Arredondo A, Chikhi L, Mazet O (2022) Heterogeneity in effective size across the genome: effects on the inverse instantaneous coalescence rate (IICR) and implications for demographic inference under linked selection. Genetics 220, iyac008. https://doi.org/10.1093/genetics/iyac008 Buffalo V (2021) Quantifying the relationship between genetic diversity and population size suggests natural selection cannot explain Lewontin's Paradox. e-Life 10, e67509. https://doi.org/10.7554/eLife.67509 Cahill JA, Soares AE, Green RE, Shapiro B (2016) Inferring species divergence times using pairwise sequential Markovian coalescent modelling and low-coverage genomic data. Philos Trans R Soc Lond B Biol Sci 371, 20150138. https://doi.org/10.1098/rstb.2015.0138 Crossman CA, Taylor EB, Barrett‐Lennard LG (2016) Hybridization in the Cetacea: widespread occurrence and associated morphological, behavioral, and ecological factors. Ecology and Evolution 6, 1293-1303. https://doi.org/10.1002/ece3.1913 Dagilis AJ, Peede D, Coughlan JM, Jofre GI, D’Agostino ERR, Mavengere H, Tate AD, Matute DR (2021) 15 years of introgression studies: quantifying gene flow across Eukaryotes. biorXiv, 2021.1106.1115.448399. https://doi.org/10.1101/2021.06.15.448399 Degnan JH, Rosenberg NA (2009) Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol Evol 24, 332-340. https://doi.org/10.1016/j.tree.2009.01.009 Durand EY, Patterson N, Reich D, Slatkin M (2011) Testing for ancient admixture between closely related populations. Mol Biol Evol 28, 2239-2252. https://doi.org/10.1093/molbev/msr048 Edelman NB, Frandsen PB, Miyagi M, Clavijo B, Davey J, Dikow RB, Garcia-Accinelli G, Van Belleghem SM, Patterson N, Neafsey DE, Challis R, Kumar S, Moreira GRP, Salazar C, Chouteau M, Counterman BA, Papa R, Blaxter M, Reed RD, Dasmahapatra KK, Kronforst M, Joron M, Jiggins CD, McMillan WO, Di Palma F, Blumberg AJ, Wakeley J, Jaffe D, Mallet J (2019) Genomic architecture and introgression shape a butterfly radiation. Science 366, 594-599. https://doi.org/10.1126/science.aaw2090 Edelman NB, Mallet J (2021) Prevalence and Adaptive Impact of Introgression. Annual Review of Genetics 55, 265-283. https://doi.org/10.1146/annurev-genet-021821-020805 Edwards CJ, Suchard MA, Lemey P, Welch JJ, Barnes I, Fulton TL, Barnett R, O'Connell TC, Coxon P, Monaghan N, Valdiosera CE, Lorenzen ED, Willerslev E, Baryshnikov GF, Rambaut A, Thomas MG, Bradley DG, Shapiro B (2011) Ancient hybridization and an Irish origin for the modern polar bear matriline. Curr Biol 21, 1251-1258. https://doi.org/10.1016/j.cub.2011.05.058 Ellegren H, Galtier N (2016) Determinants of genetic diversity. Nat Rev Genet 17, 422-433. https://doi.org/10.1038/nrg.2016.58 Flouri T, Jiao X, Rannala B, Yang Z (2018) Species Tree Inference with BPP Using Genomic Sequences and the Multispecies Coalescent. Mol Biol Evol 35, 2585-2593. https://doi.org/10.1093/molbev/msy147 Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, Hansen NF, Durand EY, Malaspinas AS, Jensen JD, Marques-Bonet T, Alkan C, Prufer K, Meyer M, Burbano HA, Good JM, Schultz R, Aximu-Petri A, Butthof A, Hober B, Hoffner B, Siegemund M, Weihmann A, Nusbaum C, Lander ES, Russ C, Novod N, Affourtit J, Egholm M, Verna C, Rudan P, Brajkovic D, Kucan Z, Gusic I, Doronichev VB, Golovanova LV, Lalueza-Fox C, de la Rasilla M, Fortea J, Rosas A, Schmitz RW, Johnson PLF, Eichler EE, Falush D, Birney E, Mullikin JC, Slatkin M, Nielsen R, Kelso J, Lachmann M, Reich D, Paabo S (2010) A draft sequence of the Neandertal genome. Science 328, 710-722. https://doi.org/10.1126/science.1188021 Guo W, Sun D, Cao Y, Xiao L, Huang X, Ren W, Xu S, Yang G (2021) Extensive Interspecific Gene Flow Shaped Complex Evolutionary History and Underestimated Species Diversity in Rapidly Radiated Dolphins. Journal of Mammalian Evolution 29, 353-367. https://doi.org/10.1007/s10914-021-09581-6 Hibbins MS, Hahn MW (2022) Phylogenomic approaches to detecting and characterizing introgression. Genetics 220, iyab173. https://doi.org/10.1093/genetics/iyab173 Jiao X, Flouri T, Rannala B, Yang Z (2020) The Impact of Cross-Species Gene Flow on Species Tree Estimation. Syst Biol 69, 830-847. https://doi.org/10.1093/sysbio/syaa001 Li H, Durbin R (2011) Inference of human population history from individual whole-genome sequences. Nature 475, 493-496. https://doi.org/10.1038/nature10231 Malinsky M, Matschiner M, Svardal H (2021) Dsuite - Fast D-statistics and related admixture evidence from VCF files. Mol Ecol Resour 21, 584-595. https://doi.org/10.1111/1755-0998.13265 Malinsky M, Svardal H, Tyers AM, Miska EA, Genner MJ, Turner GF, Durbin R (2018) Whole-genome sequences of Malawi cichlids reveal multiple radiations interconnected by gene flow. Nature Ecology & Evolution 2, 1940-1955. https://doi.org/10.1038/s41559-018-0717-x Mallet J, Besansky N, Hahn MW (2016) How reticulated are species? Bioessays 38, 140-149. https://doi.org/10.1002/bies.201500149 McGowen MR, Tsagkogeorga G, Alvarez-Carretero S, Dos Reis M, Struebig M, Deaville R, Jepson PD, Jarman S, Polanowski A, Morin PA, Rossiter SJ (2020) Phylogenomic Resolution of the Cetacean Tree of Life Using Target Sequence Capture. Syst Biol 69, 479-501. https://doi.org/10.1093/sysbio/syz068 Moura AE, Shreves K, Pilot M, Andrews KR, Moore DM, Kishida T, Möller L, Natoli A, Gaspari S, McGowen M, Chen I, Gray H, Gore M, Culloch RM, Kiani MS, Willson MS, Bulushi A, Collins T, Baldwin R, Willson A, Minton G, Ponnampalam L, Hoelzel AR (2020) Phylogenomics of the genus Tursiops and closely related Delphininae reveals extensive reticulation among lineages and provides inference about eco-evolutionary drivers. Molecular Phylogenetics and Evolution 146,107047. https://doi.org/10.1016/j.ympev.2020.106756 Müller NF, Ogilvie HA, Zhang C, Fontaine MC, Amaya-Romero JE, Drummond AJ, Stadler T (in press) Joint inference of species histories and gene flow. Syst Biol. Pease JB, Hahn MW (2015) Detection and Polarization of Introgression in a Five-Taxon Phylogeny. Syst Biol 64, 651-662. https://doi.org/10.1093/sysbio/syv023 Rabier CE, Berry V, Stoltz M, Santos JD, Wang W, Glaszmann JC, Pardi F, Scornavacca C (2021) On the inference of complex phylogenetic networks by Markov Chain Monte-Carlo. PLoS Comput Biol 17, e1008380. https://doi.org/10.1371/journal.pcbi.1008380 Seehausen O (2004) Hybridization and adaptive radiation. Trends Ecol Evol 19, 198-207. https://doi.org/10.1016/j.tree.2004.01.003 Solís-Lemus C, Yang M, Ané C (2016) Inconsistency of Species Tree Methods under Gene Flow. Syst Biol 65, 843-851. https://doi.org/10.1093/sysbio/syw030 Thawornwattana Y, Dalquen D, Yang Z, Tamura K (2018) Coalescent Analysis of Phylogenomic Data Confidently Resolves the Species Relationships in the Anopheles gambiae Species Complex. Molecular Biology and Evolution 35, 2512-2527. https://doi.org/10.1093/molbev/msy158 Wang K, Mathieson I, O’Connell J, Schiffels S (2020) Tracking human population structure through time from whole genome sequences. PLOS Genetics 16, e1008552. https://doi.org/10.1371/journal.pgen.1008552 Westbury MV, Cabrera AA, Rey-Iglesia A, Cahsan BD, Duchêne DA, Hartmann S, Lorenzen ED (2022) A genomic assessment of the marine-speciation paradox within the toothed whale superfamily Delphinoidea. bioRxiv, 2020.10.23.352286, ver. 7 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.10.23.352286 Westbury MV, Petersen B, Lorenzen ED (2019) Genomic analyses reveal an absence of contemporary introgressive admixture between fin whales and blue whales, despite known hybrids. PLoS ONE 14, e0222004. https://doi.org/10.1371/journal.pone.0222004 Yang Z (2015) The BPP program for species tree estimation and species delimitation. Current Zoology 61, 854-865. https://doi.org/10.1093/czoolo/61.5.854 Zhang C, Rabiee M, Sayyari E, Mirarab S (2018) ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19, 153. https://doi.org/10.1186/s12859-018-2129-y | A genomic assessment of the marine-speciation paradox within the toothed whale superfamily Delphinoidea | Michael V Westbury, Andrea A Cabrera, Alba Rey-Iglesia, Binia De Cahsan, David A. Duchêne, Stefanie Hartmann, Eline D Lorenzen | <p>The importance of post-divergence gene flow in speciation has been documented across a range of taxa in recent years, and may have been especially widespread in highly mobile, wide-ranging marine species, such as cetaceans. Here, we studied ind... | | Evolutionary Dynamics, Hybridization / Introgression, Molecular Evolution, Phylogenetics / Phylogenomics, Speciation | Michael C. Fontaine | 2020-10-25 08:55:50 | ||
14 Apr 2021

Parasitic success and venom composition evolve upon specialization of parasitoid wasps to different host speciesWhat makes a parasite successful? Parasitoid wasp venoms evolve rapidly in a host-specific mannerRecommended by Élio Sucena based on reviews by Simon Fellous, alexandre leitão and 1 anonymous reviewerParasitoid wasps have developed different mechanisms to increase their parasitic success, usually at the expense of host survival (Fellowes and Godfray, 2000). Eggs of these insects are deposited inside the juvenile stages of their hosts, which in turn deploy several immune response strategies to eliminate or disable them (Yang et al., 2020). Drosophila melanogaster protects itself against parasitoid attacks through the production of specific elongated haemocytes called lamellocytes which form a capsule around the invading parasite (Lavine and Strand, 2002; Rizki and Rizki, 1992) and the subsequent activation of the phenol-oxidase cascade leading to the release of toxic radicals (Nappi et al., 1995). On the parasitoid side, robust responses have evolved to evade host immune defenses as for example the Drosophila-specific endoparasite Leptopilina boulardi, which releases venom during oviposition that modifies host behaviour (Varaldi et al., 2006) and inhibits encapsulation (Gueguen et al., 2011; Martinez et al., 2012).
References Cavigliasso, F., Mathé-Hubert, H., Gatti, J.-L., Colinet, D. and Poirié, M. (2021) Parasitic success and venom composition evolve upon specialization of parasitoid wasps to different host species. bioRxiv, 2020.10.24.353417, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.10.24.353417 Cavigliasso, F., Mathé-Hubert, H., Kremmer, L., Rebuf, C., Gatti, J.-L., Malausa, T., Colinet, D., Poiré, M. and Léne. (2019). Rapid and Differential Evolution of the Venom Composition of a Parasitoid Wasp Depending on the Host Strain. Toxins, 11(629). https://doi.org/10.3390/toxins11110629 Colinet, D., Deleury, E., Anselme, C., Cazes, D., Poulain, J., Azema-Dossat, C., Belghazi, M., Gatti, J. L. and Poirié, M. (2013). Extensive inter- and intraspecific venom variation in closely related parasites targeting the same host: The case of Leptopilina parasitoids of Drosophila. Insect Biochemistry and Molecular Biology, 43(7), 601–611. https://doi.org/10.1016/j.ibmb.2013.03.010 Colinet, D., Dubuffet, A., Cazes, D., Moreau, S., Drezen, J. M. and Poirié, M. (2009). A serpin from the parasitoid wasp Leptopilina boulardi targets the Drosophila phenoloxidase cascade. Developmental and Comparative Immunology, 33(5), 681–689. https://doi.org/10.1016/j.dci.2008.11.013 Fellowes, M. D. E. and Godfray, H. C. J. (2000). The evolutionary ecology of resistance to parasitoids by Drosophila. Heredity, 84(1), 1–8. https://doi.org/10.1046/j.1365-2540.2000.00685.x Gueguen, G., Rajwani, R., Paddibhatla, I., Morales, J. and Govind, S. (2011). VLPs of Leptopilina boulardi share biogenesis and overall stellate morphology with VLPs of the heterotoma clade. Virus Research, 160(1–2), 159–165. https://doi.org/10.1016/j.virusres.2011.06.005 Lavine, M. D. and Strand, M. R. (2002). Insect hemocytes and their role in immunity. Insect Biochemistry and Molecular Biology, 32(10), 1295–1309. https://doi.org/10.1016/S0965-1748(02)00092-9 Martinez, J., Duplouy, A., Woolfit, M., Vavre, F., O’Neill, S. L. and Varaldi, J. (2012). Influence of the virus LbFV and of Wolbachia in a host-parasitoid interaction. PloS One, 7(4), e35081. https://doi.org/10.1371/journal.pone.0035081 Nappi, A. J., Vass, E., Frey, F. and Carton, Y. (1995). Superoxide anion generation in Drosophila during melanotic encapsulation of parasites. European Journal of Cell Biology, 68(4), 450–456. Poirié, M., Colinet, D. and Gatti, J. L. (2014). Insights into function and evolution of parasitoid wasp venoms. Current Opinion in Insect Science, 6, 52–60. https://doi.org/10.1016/j.cois.2014.10.004 Rizki, T. M. and Rizki, R. M. (1992). Lamellocyte differentiation in Drosophila larvae parasitized by Leptopilina. Developmental and Comparative Immunology, 16(2–3), 103–110. https://doi.org/10.1016/0145-305X(92)90011-Z Schlenke, T. A., Morales, J., Govind, S. and Clark, A. G. (2007). Contrasting infection strategies in generalist and specialist wasp parasitoids of Drosophila melanogaster. PLoS Pathogens, 3(10), 1486–1501. https://doi.org/10.1371/journal.ppat.0030158 Varaldi, J., Petit, S., Boulétreau, M. and Fleury, F. (2006). The virus infecting the parasitoid Leptopilina boulardi exerts a specific action on superparasitism behaviour. Parasitology, 132(Pt 6), 747–756. https://doi.org/10.1017/S0031182006009930 Yang, L., Qiu, L., Fang, Q., Stanley, D. W. and Gong‐Yin, Y. (2020). Cellular and humoral immune interactions between Drosophila and its parasitoids. Insect Science. https://doi.org/10.1111/1744-7917.12863
| Parasitic success and venom composition evolve upon specialization of parasitoid wasps to different host species | Fanny Cavigliasso, Hugo Mathé-Hubert, Jean-Luc Gatti, Dominique Colinet, Marylène Poirié | <p>Female endoparasitoid wasps usually inject venom into hosts to suppress their immune response and ensure offspring development. However, the parasitoid’s ability to evolve towards increased success on a given host simultaneously with the evolut... | | Experimental Evolution, Species interactions | Élio Sucena | 2020-10-26 15:00:55 | ||
12 Nov 2021

How ancient forest fragmentation and riparian connectivity generate high levels of genetic diversity in a micro-endemic Malagasy treeAn ancient age of open-canopy landscapes in northern Madagascar? Evidence from the population genetic structure of a forest treeRecommended by Miguel de Navascués based on reviews by Katharina Budde and Yurena Arjona
We currently live in the Anthropocene, the geological age characterized by a profound impact of human populations in the ecosystems and the environment. While there is little doubt about the action of humans in the shaping of present landscapes, it can be difficult to determine what the state of those landscapes was before humans started to modify them. This is the case of the Madagascar grasslands, whose origins have been debated with arguments proposing them either as anthropogenic, created with the arrival of humans around 2000BP, or as ancient features of the natural landscape with a forest fragmentation process due to environmental changes pre-dating human arrival [e.g. 1,2]. One way to clarify this question is through the genetic study of native species. Population continuity and fragmentation along time shape the structure of the genetic diversity in space. Species living in a uniform continuous habitat are expected to show genetic structuring determined only by geographical distance. Recent changes of the habitat can take many generations to reshape that genetic structure [3]. Thus, we expect genetic structure to reflect ancient features of the landscape. The work by Jordi Salmona and collaborators [4] studies the factors determining the population genetic structure of the Malagasy spiny olive (Noronhia spinifolia). This narrow endemic species is distributed in the discontinuous forest patches of the Loky-Manambato region (northern Madagascar). Jordi Salmona and collaborators genotyped 72 individuals distributed across the species distribution with restriction associated DNA sequencing and organelle microsatellite markers. Then, they studied the population genetic structure of the species. Using isolation-by-resistance models [5], they tested the influence of several landscape features (forest cover, roads, rivers, slope, etc.) on the connectivity between populations. Maternally inherited loci (chloroplast and mitochondria) and bi-parentally inherited loci (nuclear), were analysed separately in an attempt to identify the role of pollen and seed dispersal in the connectivity of populations. Despite the small distribution of the species, Jordi Salmona and collaborators [4] found remarkable levels of genetic diversity. The spatial structure of this diversity was found to be mainly explained by the forest cover of the landscape, suggesting that the landscape has been composed by patches of forests and grasslands for a long time. The main role of forest cover for the connectivity among populations also highlights the importance of riparian forest as dispersal corridors. Finally, differences between organelle and nuclear markers were not enough to establish any strong conclusion about the differences between pollen and seed dispersal. The results presented by Jordi Salmona and collaborators [4] contribute to the understanding of the history and ecology of understudied Madagascar ecosystems. Previous population genetic studies in some forest-dwelling mammals have been interpreted as supporting an old age for the fragmented landscapes in northern Madagascar [e.g. 1,6]. To my knowledge, this is the first study on a tree species. While this work might not completely settle the debate, it emphasizes the importance of studying a diversity of species to understand the biogeographic dynamics of a region. References 1. Quéméré, E., X. Amelot, J. Pierson, B. Crouau-Roy, L. Chikhi (2012) Genetic data suggest a natural prehuman origin of open habitats in northern Madagascar and question the deforestation narrative in this region. Proceedings of the National Academy of Sciences of the United States of | How ancient forest fragmentation and riparian connectivity generate high levels of genetic diversity in a micro-endemic Malagasy tree | Jordi Salmona, Axel Dresen, Anicet E. Ranaivoson, Sophie Manzi, Barbara Le Pors, Cynthia Hong-Wa, Jacqueline Razanatsoa, Nicole V. Andriaholinirina, Solofonirina Rasoloharijaona, Marie-Elodie Vavitsara, Guillaume Besnard | <p>Understanding landscape changes is central to predicting evolutionary trajectories and defining conservation practices. While human-driven deforestation is intense throughout Madagascar, exception in areas like the Loky-Manambato region (North)... | | Evolutionary Ecology, Phylogeography & Biogeography, Population Genetics / Genomics | Miguel de Navascués | 2020-11-27 09:07:21 | ||
11 May 2021
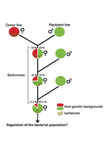
Wolbachia load variation in Drosophila is more likely caused by drift than by host genetic factorsDrift rather than host or parasite control can explain within-host Wolbachia growthRecommended by Alison Duncan and Michael Hochberg based on reviews by Simon Fellous and 1 anonymous reviewerWithin-host parasite density is tightly linked to parasite fitness often determining both transmission success and virulence (parasite-induced harm to the host) (Alizon et al., 2009, Anderson & May, 1982). Parasite density may thus be controlled by selection balancing these conflicting pressures. Actual within-host density regulation may be under host or parasite control, or due to other environmental factors (Wale et al., 2019, Vale et al., 2011, Chrostek et al., 2013). Vertically transmitted parasites may also be more vulnerable to drift associated with bottlenecks between generations, which may also determine within-host population size (Mathe-Hubert et al., 2019, Mira & Moran, 2002). Bénard et al. (2021) use 3 experiments to disentangle the role of drift versus host factors in the control of within-host Wolbachia growth in Drosophila melanogaster. They use the wMelPop Wolbachia strain in which virulence (fly longevity) and within-host growth correlate positively with copy number in the genomic region Octomom (Chrostek et al., 2013, Chrostek & Teixeira, 2015). Octomom copy number can be used as a marker for different genetic lineages within the wMelPop strain. In a first experiment, they introgressed and backcrossed this Wolbachia strain into 6 different host genetic backgrounds and show striking differences in within-host symbiont densities which correlate positively with Octomom copy number. This is consistent with host genotype selecting different Wolbachia strains, but also with bottlenecks and drift between generations. To distinguish between these possibilities, they perform 2 further experiments. A second experiment repeated experiment 1, but this time introgression was into 3 independent lines of the Bolivia and USA Drosophila populations; those that, respectively, exhibited the lowest and highest Wolbachia density and Octomom copy number. In this experiment, growth and Octomom copy number were measured across the 3 lines, for each population, after 1, 13 and 25 generations. Although there were little differences between replicates at generation 1, there were differences at generations 13 and 25 among the replicates of both the Bolivia and USA lines. These results are indicative of parasite control, or drift being responsible for within-host growth rather than host factors. A third experiment tested whether Wolbachia density and copy number were under host or parasite control. This was done, again using the USA and Bolivia lines, but this time those from the first experiment, several generations following the initial introgression and backcrossing. The newly introgressed lines were again followed for 25 generations. At generation 1, Wolbachia phenotypes resembled those of the donor parasite population and not the recipient host population indicating a possible maternal effect, but a lack of host control over the parasite. Furthermore, Wolbachia densities and Octomom number differed among replicate lines through time for Bolivia populations and from the donor parasite lines for both populations. These differences among replicate lines that share both host and parasite origins suggest that drift and/or maternal effects are responsible for within-host Wolbachia density and Octomom number. These findings indicate that drift appears to play a role in shaping Wolbachia evolution in this system. Nevertheless, completely ruling out the role of the host or parasite in controlling densities will require further study. The findings of Bénard and coworkers (2021) should stimulate future work on the contribution of drift to the evolution of vertically transmitted parasites. References Alizon S, Hurford A, Mideo N, Baalen MV (2009) Virulence evolution and the trade-off hypothesis: history, current state of affairs and the future. Journal of Evolutionary Biology, 22, 245–259. https://doi.org/10.1111/j.1420-9101.2008.01658.x Anderson RM, May RM (1982) Coevolution of hosts and parasites. Parasitology, 85, 411–426. https://doi.org/10.1017/S0031182000055360 Bénard A, Henri H, Noûs C, Vavre F, Kremer N (2021) Wolbachia load variation in Drosophila is more likely caused by drift than by host genetic factors. bioRxiv, 2020.11.29.402545, ver. 4 recommended and peer-reviewed by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.11.29.402545 Chrostek E, Marialva MSP, Esteves SS, Weinert LA, Martinez J, Jiggins FM, Teixeira L (2013) Wolbachia Variants Induce Differential Protection to Viruses in Drosophila melanogaster: A Phenotypic and Phylogenomic Analysis. PLOS Genetics, 9, e1003896. https://doi.org/10.1371/journal.pgen.1003896 Chrostek E, Teixeira L (2015) Mutualism Breakdown by Amplification of Wolbachia Genes. PLOS Biology, 13, e1002065. https://doi.org/10.1371/journal.pbio.1002065 Mathé‐Hubert H, Kaech H, Hertaeg C, Jaenike J, Vorburger C (2019) Nonrandom associations of maternally transmitted symbionts in insects: The roles of drift versus biased cotransmission and selection. Molecular Ecology, 28, 5330–5346. https://doi.org/10.1111/mec.15206 Mira A, Moran NA (2002) Estimating Population Size and Transmission Bottlenecks in Maternally Transmitted Endosymbiotic Bacteria. Microbial Ecology, 44, 137–143. https://doi.org/10.1007/s00248-002-0012-9 Vale PF, Wilson AJ, Best A, Boots M, Little TJ (2011) Epidemiological, Evolutionary, and Coevolutionary Implications of Context-Dependent Parasitism. The American Naturalist, 177, 510–521. https://doi.org/10.1086/659002 Wale N, Jones MJ, Sim DG, Read AF, King AA (2019) The contribution of host cell-directed vs. parasite-directed immunity to the disease and dynamics of malaria infections. Proceedings of the National Academy of Sciences, 116, 22386–22392. https://doi.org/10.1073/pnas.1908147116
| Wolbachia load variation in Drosophila is more likely caused by drift than by host genetic factors | Alexis Bénard, Hélène Henri, Camille Noûs, Fabrice Vavre, Natacha Kremer | <p style="text-align: justify;">Symbiosis is a continuum of long-term interactions ranging from mutualism to parasitism, according to the balance between costs and benefits for the protagonists. The density of endosymbionts is, in both cases, a ke... | | Evolutionary Dynamics, Genetic conflicts, Species interactions | Alison Duncan | 2020-12-01 16:28:14 |




