
Latest recommendations

| Id | Title | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date▲ | |
|---|---|---|---|---|---|---|---|---|---|
12 Nov 2021

How ancient forest fragmentation and riparian connectivity generate high levels of genetic diversity in a micro-endemic Malagasy treeAn ancient age of open-canopy landscapes in northern Madagascar? Evidence from the population genetic structure of a forest treeRecommended by Miguel de Navascués based on reviews by Katharina Budde and Yurena Arjona based on reviews by Katharina Budde and Yurena Arjona
We currently live in the Anthropocene, the geological age characterized by a profound impact of human populations in the ecosystems and the environment. While there is little doubt about the action of humans in the shaping of present landscapes, it can be difficult to determine what the state of those landscapes was before humans started to modify them. This is the case of the Madagascar grasslands, whose origins have been debated with arguments proposing them either as anthropogenic, created with the arrival of humans around 2000BP, or as ancient features of the natural landscape with a forest fragmentation process due to environmental changes pre-dating human arrival [e.g. 1,2]. One way to clarify this question is through the genetic study of native species. Population continuity and fragmentation along time shape the structure of the genetic diversity in space. Species living in a uniform continuous habitat are expected to show genetic structuring determined only by geographical distance. Recent changes of the habitat can take many generations to reshape that genetic structure [3]. Thus, we expect genetic structure to reflect ancient features of the landscape. The work by Jordi Salmona and collaborators [4] studies the factors determining the population genetic structure of the Malagasy spiny olive (Noronhia spinifolia). This narrow endemic species is distributed in the discontinuous forest patches of the Loky-Manambato region (northern Madagascar). Jordi Salmona and collaborators genotyped 72 individuals distributed across the species distribution with restriction associated DNA sequencing and organelle microsatellite markers. Then, they studied the population genetic structure of the species. Using isolation-by-resistance models [5], they tested the influence of several landscape features (forest cover, roads, rivers, slope, etc.) on the connectivity between populations. Maternally inherited loci (chloroplast and mitochondria) and bi-parentally inherited loci (nuclear), were analysed separately in an attempt to identify the role of pollen and seed dispersal in the connectivity of populations. Despite the small distribution of the species, Jordi Salmona and collaborators [4] found remarkable levels of genetic diversity. The spatial structure of this diversity was found to be mainly explained by the forest cover of the landscape, suggesting that the landscape has been composed by patches of forests and grasslands for a long time. The main role of forest cover for the connectivity among populations also highlights the importance of riparian forest as dispersal corridors. Finally, differences between organelle and nuclear markers were not enough to establish any strong conclusion about the differences between pollen and seed dispersal. The results presented by Jordi Salmona and collaborators [4] contribute to the understanding of the history and ecology of understudied Madagascar ecosystems. Previous population genetic studies in some forest-dwelling mammals have been interpreted as supporting an old age for the fragmented landscapes in northern Madagascar [e.g. 1,6]. To my knowledge, this is the first study on a tree species. While this work might not completely settle the debate, it emphasizes the importance of studying a diversity of species to understand the biogeographic dynamics of a region. References 1. Quéméré, E., X. Amelot, J. Pierson, B. Crouau-Roy, L. Chikhi (2012) Genetic data suggest a natural prehuman origin of open habitats in northern Madagascar and question the deforestation narrative in this region. Proceedings of the National Academy of Sciences of the United States of | How ancient forest fragmentation and riparian connectivity generate high levels of genetic diversity in a micro-endemic Malagasy tree | Jordi Salmona, Axel Dresen, Anicet E. Ranaivoson, Sophie Manzi, Barbara Le Pors, Cynthia Hong-Wa, Jacqueline Razanatsoa, Nicole V. Andriaholinirina, Solofonirina Rasoloharijaona, Marie-Elodie Vavitsara, Guillaume Besnard | <p>Understanding landscape changes is central to predicting evolutionary trajectories and defining conservation practices. While human-driven deforestation is intense throughout Madagascar, exception in areas like the Loky-Manambato region (North)... | | Evolutionary Ecology, Phylogeography & Biogeography, Population Genetics / Genomics | Miguel de Navascués | 2020-11-27 09:07:21 | ||
11 May 2021
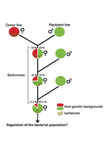
Wolbachia load variation in Drosophila is more likely caused by drift than by host genetic factorsDrift rather than host or parasite control can explain within-host Wolbachia growthRecommended by Alison Duncan and Michael Hochberg based on reviews by Simon Fellous and 1 anonymous reviewerWithin-host parasite density is tightly linked to parasite fitness often determining both transmission success and virulence (parasite-induced harm to the host) (Alizon et al., 2009, Anderson & May, 1982). Parasite density may thus be controlled by selection balancing these conflicting pressures. Actual within-host density regulation may be under host or parasite control, or due to other environmental factors (Wale et al., 2019, Vale et al., 2011, Chrostek et al., 2013). Vertically transmitted parasites may also be more vulnerable to drift associated with bottlenecks between generations, which may also determine within-host population size (Mathe-Hubert et al., 2019, Mira & Moran, 2002). Bénard et al. (2021) use 3 experiments to disentangle the role of drift versus host factors in the control of within-host Wolbachia growth in Drosophila melanogaster. They use the wMelPop Wolbachia strain in which virulence (fly longevity) and within-host growth correlate positively with copy number in the genomic region Octomom (Chrostek et al., 2013, Chrostek & Teixeira, 2015). Octomom copy number can be used as a marker for different genetic lineages within the wMelPop strain. In a first experiment, they introgressed and backcrossed this Wolbachia strain into 6 different host genetic backgrounds and show striking differences in within-host symbiont densities which correlate positively with Octomom copy number. This is consistent with host genotype selecting different Wolbachia strains, but also with bottlenecks and drift between generations. To distinguish between these possibilities, they perform 2 further experiments. A second experiment repeated experiment 1, but this time introgression was into 3 independent lines of the Bolivia and USA Drosophila populations; those that, respectively, exhibited the lowest and highest Wolbachia density and Octomom copy number. In this experiment, growth and Octomom copy number were measured across the 3 lines, for each population, after 1, 13 and 25 generations. Although there were little differences between replicates at generation 1, there were differences at generations 13 and 25 among the replicates of both the Bolivia and USA lines. These results are indicative of parasite control, or drift being responsible for within-host growth rather than host factors. A third experiment tested whether Wolbachia density and copy number were under host or parasite control. This was done, again using the USA and Bolivia lines, but this time those from the first experiment, several generations following the initial introgression and backcrossing. The newly introgressed lines were again followed for 25 generations. At generation 1, Wolbachia phenotypes resembled those of the donor parasite population and not the recipient host population indicating a possible maternal effect, but a lack of host control over the parasite. Furthermore, Wolbachia densities and Octomom number differed among replicate lines through time for Bolivia populations and from the donor parasite lines for both populations. These differences among replicate lines that share both host and parasite origins suggest that drift and/or maternal effects are responsible for within-host Wolbachia density and Octomom number. These findings indicate that drift appears to play a role in shaping Wolbachia evolution in this system. Nevertheless, completely ruling out the role of the host or parasite in controlling densities will require further study. The findings of Bénard and coworkers (2021) should stimulate future work on the contribution of drift to the evolution of vertically transmitted parasites. References Alizon S, Hurford A, Mideo N, Baalen MV (2009) Virulence evolution and the trade-off hypothesis: history, current state of affairs and the future. Journal of Evolutionary Biology, 22, 245–259. https://doi.org/10.1111/j.1420-9101.2008.01658.x Anderson RM, May RM (1982) Coevolution of hosts and parasites. Parasitology, 85, 411–426. https://doi.org/10.1017/S0031182000055360 Bénard A, Henri H, Noûs C, Vavre F, Kremer N (2021) Wolbachia load variation in Drosophila is more likely caused by drift than by host genetic factors. bioRxiv, 2020.11.29.402545, ver. 4 recommended and peer-reviewed by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.11.29.402545 Chrostek E, Marialva MSP, Esteves SS, Weinert LA, Martinez J, Jiggins FM, Teixeira L (2013) Wolbachia Variants Induce Differential Protection to Viruses in Drosophila melanogaster: A Phenotypic and Phylogenomic Analysis. PLOS Genetics, 9, e1003896. https://doi.org/10.1371/journal.pgen.1003896 Chrostek E, Teixeira L (2015) Mutualism Breakdown by Amplification of Wolbachia Genes. PLOS Biology, 13, e1002065. https://doi.org/10.1371/journal.pbio.1002065 Mathé‐Hubert H, Kaech H, Hertaeg C, Jaenike J, Vorburger C (2019) Nonrandom associations of maternally transmitted symbionts in insects: The roles of drift versus biased cotransmission and selection. Molecular Ecology, 28, 5330–5346. https://doi.org/10.1111/mec.15206 Mira A, Moran NA (2002) Estimating Population Size and Transmission Bottlenecks in Maternally Transmitted Endosymbiotic Bacteria. Microbial Ecology, 44, 137–143. https://doi.org/10.1007/s00248-002-0012-9 Vale PF, Wilson AJ, Best A, Boots M, Little TJ (2011) Epidemiological, Evolutionary, and Coevolutionary Implications of Context-Dependent Parasitism. The American Naturalist, 177, 510–521. https://doi.org/10.1086/659002 Wale N, Jones MJ, Sim DG, Read AF, King AA (2019) The contribution of host cell-directed vs. parasite-directed immunity to the disease and dynamics of malaria infections. Proceedings of the National Academy of Sciences, 116, 22386–22392. https://doi.org/10.1073/pnas.1908147116
| Wolbachia load variation in Drosophila is more likely caused by drift than by host genetic factors | Alexis Bénard, Hélène Henri, Camille Noûs, Fabrice Vavre, Natacha Kremer | <p style="text-align: justify;">Symbiosis is a continuum of long-term interactions ranging from mutualism to parasitism, according to the balance between costs and benefits for the protagonists. The density of endosymbionts is, in both cases, a ke... | | Evolutionary Dynamics, Genetic conflicts, Species interactions | Alison Duncan | 2020-12-01 16:28:14 | ||
26 Oct 2021

Large-scale geographic survey provides insights into the colonization history of a major aphid pest on its cultivated apple host in Europe, North America and North AfricaThe evolutionary puzzle of the host-parasite-endosymbiont Russian doll for apples and aphidsRecommended by Ignacio Bravo based on reviews by Pedro Simões and 1 anonymous reviewerEach individual multicellular organism, each of our bodies, is a small universe. Every living surface -skin, cuticle, bark, mucosa- is the home place to milliards of bacteria, fungi and viruses. They constitute our microbiota. Some of them are essential for certain organisms. Other could not live without their hosts. For many species, the relationship between host and microbiota is so close that their histories are inseparable. The recognition of this biological inextricability has led to the notion of holobiont as the organism ensemble of host and microbiota. When individuals of a particular animal or plant species expand their geographical range, it is the holobiont that expands. And these processes of migration, expansion and colonization are often accompanied by evolutionary and ecological innovations in the interspecies relationships, at the macroscopic level (e.g. novel predator-prey or host-parasite interactions) and at the microscopic level (e.g. changes in the microbiota composition). From the human point of view, these novel interactions can be economically disastrous if they involve and threaten important crop or cattle species. And this is especially worrying in the present context of genetic standardization and intensification for mass-production on the one hand, and of climate change on the other. With this perspective, the international team led by Amandine Cornille presents a study aiming at understanding the evolutionary history of the rosy apple aphid Dysaphis plantaginea Passerini, a major pest of the cultivated apple tree Malus domestica Borkh (1). The apple tree was probably domesticated in Central Asia, and later disseminated by humans over the world in different waves, and it was probably introduced in Europe by the Greeks. It is however unclear when and where D. plantaginea started parasitizing the cultivated apple tree. The ancestral D. plantaginea could have already infected the wild ancestor of current cultivated apple trees, but the aphid is not common in Central Asia. Alternatively, it may have gained access only later to the plant, possibly via a host jump, from Pyrus to Malus that may have occurred in Asia Minor or in the Caucasus. In the present preprint, Olvera-Vázquez and coworkers have analysed over 650 D. plantaginea colonies from 52 orchards in 13 countries, in Western, Central and Eastern Europe as well as in Morocco and the USA. The authors have analysed the genetic diversity in the sampled aphids, and have characterized as well the composition of the associated endosymbiont bacteria. The analyses detect substantial recent admixture, but allow to identify aphid subpopulations slightly but significantly differentiated and isolated by distance, especially those in Morocco and the USA, as well as to determine the presence of significant gene flow. This process of colonization associated to gene flow is most likely indirectly driven by human interactions. Very interestingly, the data show that this genetic diversity in the aphids is not reflected by a corresponding diversity in the associated microbiota, largely dominated by a few Buchnera aphidicola variants. In order to determine polarity in the evolutionary history of the aphid-tree association, the authors have applied approximate Bayesian computing and machine learning approaches. Albeit promising, the results are not sufficiently robust to assess directionality nor to confidently assess the origin of the crop pest. Despite the large effort here communicated, the authors point to the lack of sufficient data (in terms of aphid isolates), especially originating from Central Asia. Such increased sampling will need to be implemented in the future in order to elucidate not only the origin and the demographic history of the interaction between the cultivated apple tree and the rosy apple aphid. This knowledge is needed to understand how this crop pest struggles with the different seasonal and geographical selection pressures while maintaining high genetic diversity, conspicuous gene flow, differentiated populations and low endosymbiontic diversity. References
| Large-scale geographic survey provides insights into the colonization history of a major aphid pest on its cultivated apple host in Europe, North America and North Africa | Olvera-Vazquez S.G., Remoué C., Venon A, Rousselet A., Grandcolas O., Azrine M., Momont L., Galan M., Benoit L., David G., Alhmedi A., Beliën T., Alins G., Franck P., Haddioui A., Jacobsen S.K., Andreev R., Simon S., Sigsgaard L., Guibert E., Tour... | <p style="text-align: justify;">With frequent host shifts involving the colonization of new hosts across large geographical ranges, crop pests are good models for examining the mechanisms of rapid colonization. The microbial partners of pest insec... | | Phylogeography & Biogeography, Population Genetics / Genomics, Species interactions | Ignacio Bravo | 2020-12-11 19:22:54 | ||
31 Mar 2022
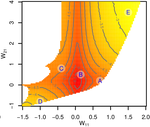
Gene network robustness as a multivariate characterGenetic and environmental robustness are distinct yet correlated evolvable traits in a gene networkRecommended by Frédéric Guillaume based on reviews by Diogo Melo, Charles Mullon and Charles Rocabert
Organisms often show robustness to genetic or environmental perturbations. Whether these two components of robustness can evolve separately is the focus of the paper by Le Rouzic [1]. Using theoretical analysis and individual-based computer simulations of a gene regulatory network model, he shows that multiple aspects of robustness can be investigated as a set of pleiotropically linked quantitative traits. While genetically correlated, various robustness components (e.g., mutational, developmental, homeostasis) of gene expression in the regulatory network evolved more or less independently from each other under directional selection. The quantitative approach of Le Rouzic could explain both how unselected robustness components can respond to selection on other components and why various robustness-related features seem to have their own evolutionary history. Moreover, he shows that all components were evolvable, but not all to the same extent. Robustness to environmental disturbances and gene expression stability showed the largest responses while increased robustness to genetic disturbances was slower. Interestingly, all components were positively correlated and remained so after selection for increased or decreased robustness. This study is an important contribution to the discussion of the evolution of robustness in biological systems. While it has long been recognized that organisms possess the ability to buffer genetic and environmental perturbations to maintain homeostasis (e.g., canalization [2]), the genetic basis and evolutionary routes to robustness and canalization are still not well understood. Models of regulatory gene networks have often been used to address aspects of robustness evolution (e.g., [3]). Le Rouzic [1] used a gene regulatory network model derived from Wagner’s model [4]. The model has as end product the expression level of a set of genes influenced by a set of regulatory elements (e.g., transcription factors). The level and stability of expression are a property of the regulatory interactions in the network. Le Rouzic made an important contribution to the study of such gene regulation models by using a quantitative genetics approach to the evolution of robustness. He crafted a way to assess the mutational variability and selection response of the components of robustness he was interested in. Le Rouzic’s approach opens avenues to investigate further aspects of gene network evolutionary properties, for instance to understand the evolution of phenotypic plasticity. Le Rouzic also discusses ways to measure his different robustness components in empirical studies. As the model is about gene expression levels at a set of protein-coding genes influenced by a set of regulatory elements, it naturally points to the possibility of using RNA sequencing to measure the variation of gene expression in know gene networks and assess their robustness. Robustness could then be studied as a multidimensional quantitative trait in an experimental setting. References [1] Le Rouzic, A (2022) Gene network robustness as a multivariate character. arXiv: 2101.01564, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://arxiv.org/abs/2101.01564 [2] Waddington CH (1942) Canalization of Development and the Inheritance of Acquired Characters. Nature, 150, 563–565. https://doi.org/10.1038/150563a0 [3] Draghi J, Whitlock M (2015) Robustness to noise in gene expression evolves despite epistatic constraints in a model of gene networks. Evolution, 69, 2345–2358. https://doi.org/10.1111/evo.12732 [4] Wagner A (1994) Evolution of gene networks by gene duplications: a mathematical model and its implications on genome organization. Proceedings of the National Academy of Sciences, 91, 4387–4391. https://doi.org/10.1073/pnas.91.10.4387 | Gene network robustness as a multivariate character | Arnaud Le Rouzic | <p style="text-align: justify;">Robustness to genetic or environmental disturbances is often considered as a key property of living systems. Yet, in spite of being discussed since the 1950s, how robustness emerges from the complexity of genetic ar... | | Bioinformatics & Computational Biology, Evolutionary Theory, Genotype-Phenotype, Quantitative Genetics | Frédéric Guillaume | Charles Mullon, Charles Rocabert, Diogo Melo | 2021-01-11 17:48:20 | |
11 Oct 2022
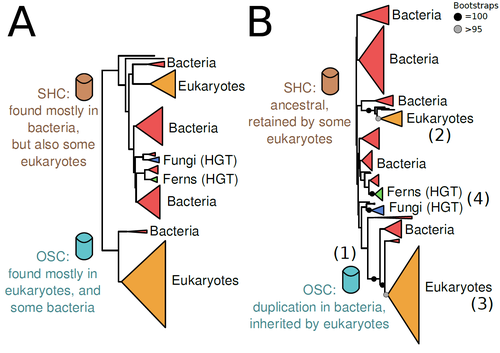
The Eukaryotic Last Common Ancestor Was Bifunctional for Hopanoid and Sterol ProductionGene family analysis suggests new evolutionary scenario for sterol and hopanoid biomarkersRecommended by Iker Irisarri based on reviews by Samuel Abalde, Denis Baurain and Jose Ramon Pardos-BlasSterols and hopanoids are sometimes used as biomarkers to infer the origin of certain groups of organisms. Traditionally, hopanoid-derived products in ancient rocks have been considered to indicate the presence of bacteria, whereas sterol derivatives have been considered to be exclusive to eukaryotes. However, a closer look at the topic reveals a rather complex distribution of either compound in both bacteria and eukaryotes. (1). The known biosynthetic pathways for sterols and hopanoids are similar but diverge at a critical step where two different enzymes are used: squalene-hopene cyclase (SHC) and oxidosqualene cyclase (OSC), the latter requiring oxygen. These two enzymes belong to the same gene family, whose complex evolutionary history is difficult to reconcile with the known species phylogeny. In this study (2), Dr. Warren R. Francis revisits the evolution of this gene family using an extended dataset with a broader taxonomic representation. In contrast to the traditional representation of the tree rooted between SHC and OSC paralogs (i.e., based on function), the author proposes that rooting the tree within bacterial SHCs and assuming a secondary origin of OSC is more parsimonious. This postulates SHC to be the ancestral function –retained in many extant bacteria and some eukaryotes– and OSC to have emerged later within bacteria –currently being mostly present in eukaryotes–. The reconstructed evolutionary history is arguably complex and can only be reconciled with the species' phylogeny by invoking many secondary losses. These losses are considered likely because many extant species acquire sterols and hopanoids by diet and lack one or both enzymes. Some cases of recent horizontal gene transfer are also proposed. In contrast to the dichotomy between bacterial SHCs and eukaryote OSCs, the new proposed scenario suggests that the eukaryote ancestor likely inherited both enzymes from bacteria and thus could be able to synthesize both sterols and hopanoids. Under this hypothesis, not only bacteria but also eukaryotes could be responsible for the hopane found in old rocks. This agrees with eukaryote fossils dating back to more than 1 billion years ago (3). Also, the observed increase of sterane levels in rocks ~600-700 million years old cannot be associated with the origin of eukaryotes, which is a much older event, but could rather reflect changes in atmospheric oxygen levels because oxygen is required for the synthesis of sterols by OSC. References 1. Santana-Molina C, Rivas-Marin E, Rojas AM, Devos DP (2020) Origin and Evolution of Polycyclic Triterpene Synthesis. Molecular Biology and Evolution, 37, 1925–1941. https://doi.org/10.1093/molbev/msaa054 2. Francis WR (2022) The Eukaryotic Last Common Ancestor Was Bifunctional for Hopanoid and Sterol Production. Preprints, 2020040186, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.20944/preprints202004.0186.v5 3. Butterfield NJ (2000) Bangiomorpha pubescens n. gen., n. sp.: implications for the evolution of sex, multicellularity, and the Mesoproterozoic/Neoproterozoic radiation of eukaryotes. Paleobiology, 26, 386–404. https://doi.org/10.1666/0094-8373(2000)026<0386:BPNGNS>2.0.CO;2 | The Eukaryotic Last Common Ancestor Was Bifunctional for Hopanoid and Sterol Production | Warren R Francis | <p>Steroid and hopanoid biomarkers can be found in ancient rocks and may give a glimpse of what life was present at that time. Sterols and hopanoids are produced by two related enzymes, though the evolutionary history of this protein family is com... | | Bioinformatics & Computational Biology, Evolutionary Ecology, Molecular Evolution, Paleontology, Phylogenetics / Phylogenomics | Iker Irisarri | 2021-01-13 16:03:29 | ||
01 Sep 2021

Connectivity and selfing drives population genetic structure in a patchy landscape: a comparative approach of four co-occurring freshwater snail speciesDeterminants of population genetic structure in co-occurring freshwater snailsRecommended by Trine Bilde and Matteo Fumagalli based on reviews by 3 anonymous reviewers
Genetic diversity is a key aspect of biodiversity and has important implications for evolutionary potential and thereby the persistence of species. Improving our understanding of the factors that drive genetic structure within and between populations is, therefore, a long-standing goal in evolutionary biology. However, this is a major challenge, because of the complex interplay between genetic drift, migration, and extinction/colonization dynamics on the one hand, and the biology and ecology of species on the other hand (Romiguier et al. 2014, Ellegren and Galtier 2016, Charlesworth 2003). Jarne et al. (2021) studied whether environmental and demographic factors affect the population genetic structure of four species of hermaphroditic freshwater snails in a similar way, using comparative analyses of neutral genetic microsatellite markers. Specifically, they investigated microsatellite variability of Hygrophila in almost 280 sites in Guadeloupe, Lesser Antilles, as part of a long-term survey experiment (Lamy et al. 2013). They then modelled the influence of the mating system, local environmental characteristics and demographic factors on population genetic diversity. Consistent with theoretical predictions (Charlesworth 2003), they detected higher genetic variation in two outcrossing species than in two selfing species, emphasizing the importance of the mating system in maintaining genetic diversity. The study further identified an important role of site connectivity, through its influences on effective population size and extinction/colonisation events. Finally, the study detects an influence of interspecific interactions caused by an ongoing invasion by one of the studied species on genetic structure, highlighting the indirect effect of changes in community composition and demography on population genetics. Jarne et al. (2021) could address the extent to which genetic structure is determined by demographic and environmental factors in multiple species given the remarkable sampling available. Additionally, the study system is extremely suitable to address this hypothesis as species’ habitats are defined and delineated. Whilst the authors did attempt to test for across-species correlations, further investigations on this matter are required. Moreover, the effect of interactions between factors should be appropriately considered in any modelling between genetic structure and local environmental or demographic features. The findings in this study contribute to improving our understanding of factors influencing population genetic diversity, and highlights the complexity of interacting factors, therefore also emphasizing the challenges of drawing general implications, additionally hampered by the relatively limited number of species studied. Jarne et al. (2021) provide an excellent showcase of an empirical framework to test determinants of genetic structure in natural populations. As such, this study can be an example for further attempts of comparative analysis of genetic diversity. References Charlesworth, D. (2003) Effects of inbreeding on the genetic diversity of populations. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences, 358, 1051-1070. doi: https://doi.org/10.1098/rstb.2003.1296 Ellegren, H. and Galtier, N. (2016) Determinants of genetic diversity. Nature Reviews Genetics, 17, 422-433. doi: https://doi.org/10.1038/nrg.2016.58 Jarne, P., Lozano del Campo, A., Lamy, T., Chapuis, E., Dubart, M., Segard, A., Canard, E., Pointier, J.-P. and David, P. (2021) Connectivity and selfing drives population genetic structure in a patchy landscape: a comparative approach of four co-occurring freshwater snail species. HAL, hal-03295242, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://hal.archives-ouvertes.fr/hal-03295242 Lamy, T., Gimenez, O., Pointier, J. P., Jarne, P. and David, P. (2013). Metapopulation dynamics of species with cryptic life stages. The American Naturalist, 181, 479-491. doi: https://doi.org/10.1086/669676 Romiguier, J., Gayral, P., Ballenghien, M. et al. (2014) Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature, 515, 261-263. doi: https://doi.org/10.1038/nature13685 | Connectivity and selfing drives population genetic structure in a patchy landscape: a comparative approach of four co-occurring freshwater snail species | Jarne P., Lozano del Campo A., Lamy T., Chapuis E., Dubart M., Segard A., Canard E., Pointier J.-P., David P. | <p style="text-align: justify;">The distribution of neutral genetic variation in subdivided populations is driven by the interplay between genetic drift, migration, local extinction and colonization. The influence of environmental and demographic ... | | Adaptation, Evolutionary Dynamics, Population Genetics / Genomics, Reproduction and Sex, Species interactions | Trine Bilde | 2021-02-11 19:57:51 | ||
30 Aug 2021
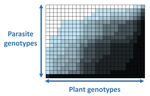
The quasi-universality of nestedness in the structure of quantitative plant-parasite interactionsNestedness and modularity in plant-parasite infection networksRecommended by Santiago Elena based on reviews by Rubén González and 2 anonymous reviewers
In a landmark paper, Flores et al. (2011) showed that the interactions between bacteria and their viruses could be nicely described using a bipartite infection networks. Two quantitative properties of these networks were of particular interest, namely modularity and nestedness. Modularity emerges when groups of host species (or genotypes) shared groups of viruses. Nestedness provided a view of the degree of specialization of both partners: high nestedness suggests that hosts differ in their susceptibility to infection, with some highly susceptible host genotypes selecting for very specialized viruses while strongly resistant host genotypes select for generalist viruses. Translated to the plant pathology parlance, this extreme case would be equivalent to a gene-for-gene infection model (Flor 1956): new mutations confer hosts with resistance to recently evolved viruses while maintaining resistance to past viruses. Likewise, virus mutations for expanding host range evolve without losing the ability to infect ancestral host genotypes. By contrast, a non-nested network would represent a matching-allele infection model (Frank 2000) in which each interacting organism evolves by losing its capacity to resist/infect its ancestral partners, resembling a Red Queen dynamic. Obviously, the reality is more complex and may lie anywhere between these two extreme situations. Recently, Valverde et al. (2020) developed a model to explain the emergence of nestedness and modularity in plant-virus infection networks across diverse habitats. They found that local modularity could coexist with global nestedness and that intraspecific competition was the main driver of the evolution of ecosystems in a continuum between nested-modular and nested networks. These predictions were tested with field data showing the association between plant host species and different viruses in different agroecosystems (Valverde et al. 2020). The effect of interspecific competition in the structure of empirical plant host-virus infection networks was also tested by McLeish et al. (2019). Besides data from agroecosystems, evolution experiments have also shown the pervasive emergence of nestedness during the diversification of independently-evolved lineages of potyviruses in Arabidopsis thaliana genotypes that differ in their susceptibility to infection (Hillung et al. 2014; González et al. 2019; Navarro et al. 2020). In their study, Moury et al. (2021) have expanded all these previous observations to a diverse set of pathosystems that range from viruses, bacteria, oomycetes, fungi, nematodes to insects. While modularity was barely seen in only a few of the systems, nestedness was a common trend (observed in ~94% of all systems). This nestedness, as seen in previous studies and as predicted by theory, emerged as a consequence of the existence of generalist and specialist strains of the parasites that differed in their capacity to infect more or less resistant plant genotypes. As pointed out by Moury et al. (2021) in their conclusions, the ubiquity of nestedness in plant-parasite infection matrices has strong implications for the evolution and management of infectious diseases. References Flor, H. H. (1956). The complementary genic systems in flax and flax rust. In Advances in genetics, 8, 29-54. https://doi.org/10.1016/S0065-2660(08)60498-8 Flores, C. O., Meyer, J. R., Valverde, S., Farr, L., and Weitz, J. S. (2011). Statistical structure of host–phage interactions. Proceedings of the National Academy of Sciences, 108, E288-E297. https://doi.org/10.1073/pnas.1101595108 Frank, S. A. (2000). Specific and non-specific defense against parasitic attack. Journal of Theoretical Biology, 202, 283-304. https://doi.org/10.1006/jtbi.1999.1054 González, R., Butković, A., and Elena, S. F. (2019). Role of host genetic diversity for susceptibility-to-infection in the evolution of virulence of a plant virus. Virus evolution, 5(2), vez024. https://doi.org/10.1093/ve/vez052 Hillung, J., Cuevas, J. M., Valverde, S., and Elena, S. F. (2014). Experimental evolution of an emerging plant virus in host genotypes that differ in their susceptibility to infection. Evolution, 68, 2467-2480. https://doi.org/10.1111/evo.12458 McLeish, M., Sacristán, S., Fraile, A., and García-Arenal, F. (2019). Coinfection organizes epidemiological networks of viruses and hosts and reveals hubs of transmission. Phytopathology, 109, 1003-1010. https://doi.org/10.1094/PHYTO-08-18-0293-R Moury B, Audergon J-M, Baudracco-Arnas S, Krima SB, Bertrand F, Boissot N, Buisson M, Caffier V, Cantet M, Chanéac S, Constant C, Delmotte F, Dogimont C, Doumayrou J, Fabre F, Fournet S, Grimault V, Jaunet T, Justafré I, Lefebvre V, Losdat D, Marcel TC, Montarry J, Morris CE, Omrani M, Paineau M, Perrot S, Pilet-Nayel M-L and Ruellan Y (2021) The quasi-universality of nestedness in the structure of quantitative plant-parasite interactions. bioRxiv, 2021.03.03.433745, ver. 4 recommended and peer-reviewed by PCI Evolutionary Biology. https://doi.org/10.1101/2021.03.03.433745 Navarro, R., Ambros, S., Martinez, F., Wu, B., Carrasco, J. L., and Elena, S. F. (2020). Defects in plant immunity modulate the rates and patterns of RNA virus evolution. bioRxiv. doi: https://doi.org/10.1101/2020.10.13.337402 Valverde, S., Vidiella, B., Montañez, R., Fraile, A., Sacristán, S., and García-Arenal, F. (2020). Coexistence of nestedness and modularity in host–pathogen infection networks. Nature ecology & evolution, 4, 568-577. https://doi.org/10.1038/s41559-020-1130-9 | The quasi-universality of nestedness in the structure of quantitative plant-parasite interactions | Moury Benoît, Audergon Jean-Marc, Baudracco-Arnas Sylvie, Ben Krima Safa, Bertrand François, Boissot Nathalie, Buisson Mireille, Caffier Valérie, Cantet Mélissa, Chanéac Sylvia, Constant Carole, Delmotte François, Dogimont Catherine, Doumayrou Jul... | <p>Understanding the relationships between host range and pathogenicity for parasites, and between the efficiency and scope of immunity for hosts are essential to implement efficient disease control strategies. In the case of plant parasites, most... | | Bioinformatics & Computational Biology, Evolutionary Dynamics, Species interactions | Santiago Elena | 2021-03-04 21:23:08 | ||
11 Oct 2021

Landscape connectivity alters the evolution of density-dependent dispersal during pushed range expansionsPhenotypic evolution during range expansions is contingent on connectivity and density dependenceRecommended by Inês Fragata based on reviews by 3 anonymous reviewersUnderstanding the mechanisms underlying range expansions is key for predicting species distributions in response to environmental changes (such as global warming) and managing the global expansion of invasive species (Parmesan 2006; Suarez & Tsutsui 2008). Traditionally, two types of ecological processes were studied as essential in shaping range expansion: dispersal and population growth. However, ecology and evolution are intertwined in range expansions, as phenotypic evolution of traits involved in demographic and dispersal patterns and processes can affect and be affected by ecological dynamics, representing a full eco-evolutionary loop (Williams et al. 2019; Miller et al. 2020). Range expansions can be characterized by the type of population growth and dispersal, divided into pushed or pulled range expansions. Species that have high dispersal and high population growth at low densities present pulled range expansions (pulled by individuals from the edge populations). In contrast, populations presenting increased growth rate at intermediate densities (due to Allee effects - Allee & Bowen 1932; i.e. where growth rate decreases at lower densities) and high dispersal at high densities present pushed range expansions (driven by individuals from core and intermediate populations) (Gandhi et al. 2016). Importantly, the type of expansion is expected to have very different consequences on the genetic (and therefore) phenotypic composition of core and edge populations. Specifically, genetic variability is expected to be lower in populations experiencing pulled expansions and higher in populations involved in pushed expansions (Gandhi et al. 2016; Miller et al. 2020). However, it is not always possible to distinguish between pulled and pushed expansions, as variation in speed and shape can overlap between the two types. In addition, it is difficult to experimentally manipulate the strength of the Allee effect to create pushed versus pulled expansions. Thus, several critical predictions regarding the genetic and phenotypic composition of pulled and pushed expansions are lacking empirical tests (but see Gandhi et al. 2016). In a previous study, Dahirel et al. (2021a) combined simulations and experimental evolution of the small wasps Trichogramma brassicae to show that low connectivity led to more pushed expansions, and higher connectivity generated more pulled expansions. In accordance with theoretical predictions, this led to reduced genetic diversity in pulled expansions, and the reverse pattern in pushed expansions. However, the question of how pulled and pushed expansions affect trait evolution remained unanswered. In this follow-up study, Dahirel et al. (2021b) tackled this issue and linked the changes in connectivity and type of expansion with the phenotypic evolution of several traits using individuals from their previous experiment. Namely, the authors compared core and edge populations with founder strains to test how evolution in pushed vs. pulled expansions affected wasp size, short movement, fecundity, dispersal, and density dependent dispersal. When density dependence was not accounted for, phenotypic changes in edge populations did not match the expectations from changes in expansion dynamics. This could be due to genetic trade-offs between traits that limit phenotypic evolution (Urquhart & Williams 2021). However, when accounting for density dependent dispersal, Dahirel et al. (2021b) observed that more connected landscapes (with pulled expansions) showed positive density dispersal in core populations and negative density dispersal in edge populations, similarly to other studies (e.g. Fronhofer et al. 2017). Interestingly, in pushed (with lower connectivity) landscapes, such shift was not observed. Instead, edge populations maintained positive density dispersal even after 14 generations of expansion, whereas core populations showed higher dispersal at lower density. The authors suggest that this seemingly contradictory result is due to a combination of three processes: 1) the expansion reduced positive density dispersal in edge populations; 2) reduced connectivity directly increased dispersal costs, increasing high density dispersal; and 3) reduced connectivity indirectly caused demographic stochasticity (and reduced temporal variability in patches) leading to higher dispersal at low density in core populations. However, these results must be taken with a grain of salt, since only one of the four experimental replicates were used in the density dependent dispersal experiment. In range expansions experiments, replication is fundamental, since stochastic processes (such as gene surfing, where alleles maybe rise in frequency due by chance) are prevalent (Miller et al. 2020), and results are highly dependent on sample size, or number of replicate populations analysed. Having said that, results from Dahirel et al. (2021b) highlight the importance to contextualize the management of invasions and species distribution, since it is thought that pulled expansions are more prevalent in nature, but pushed expansions can be more important in scenarios where patchiness is high, such as urban landscapes. Moreover, Dahirel's et al. (2021b) study is a first step showing that accounting for trait density dependence is crucial when following phenotypic evolution during range expansion, and that evolution of density dependent traits may be constrained by landscape conditions. This highlights the need to account for both connectivity and density dependence to draw more accurate predictions on the evolutionary and ecological outcomes of range expansions. Allee WC, Bowen ES (1932) Studies in animal aggregations: Mass protection against colloidal silver among goldfishes. Journal of Experimental Zoology, 61, 185–207. https://doi.org/10.1002/jez.1400610202 Dahirel M, Bertin A, Calcagno V, Duraj C, Fellous S, Groussier G, Lombaert E, Mailleret L, Marchand A, Vercken E (2021a) Landscape connectivity alters the evolution of density-dependent dispersal during pushed range expansions. bioRxiv, 2021.03.03.433752, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.03.03.433752 Dahirel M, Bertin A, Haond M, Blin A, Lombaert E, Calcagno V, Fellous S, Mailleret L, Malausa T, Vercken E (2021b) Shifts from pulled to pushed range expansions caused by reduction of landscape connectivity. Oikos, 130, 708–724. https://doi.org/10.1111/oik.08278 Fronhofer EA, Gut S, Altermatt F (2017) Evolution of density-dependent movement during experimental range expansions. Journal of Evolutionary Biology, 30, 2165–2176. https://doi.org/10.1111/jeb.13182 Gandhi SR, Yurtsev EA, Korolev KS, Gore J (2016) Range expansions transition from pulled to pushed waves as growth becomes more cooperative in an experimental microbial population. Proceedings of the National Academy of Sciences, 113, 6922–6927. https://doi.org/10.1073/pnas.1521056113 Miller TEX, Angert AL, Brown CD, Lee-Yaw JA, Lewis M, Lutscher F, Marculis NG, Melbourne BA, Shaw AK, Szűcs M, Tabares O, Usui T, Weiss-Lehman C, Williams JL (2020) Eco-evolutionary dynamics of range expansion. Ecology, 101, e03139. https://doi.org/10.1002/ecy.3139 Parmesan C (2006) Ecological and Evolutionary Responses to Recent Climate Change. Annual Review of Ecology, Evolution, and Systematics, 37, 637–669. https://doi.org/10.1146/annurev.ecolsys.37.091305.110100 Suarez AV, Tsutsui ND (2008) The evolutionary consequences of biological invasions. Molecular Ecology, 17, 351–360. https://doi.org/10.1111/j.1365-294X.2007.03456.x Urquhart CA, Williams JL (2021) Trait correlations and landscape fragmentation jointly alter expansion speed via evolution at the leading edge in simulated range expansions. Theoretical Ecology. https://doi.org/10.1007/s12080-021-00503-z Williams JL, Hufbauer RA, Miller TEX (2019) How Evolution Modifies the Variability of Range Expansion. Trends in Ecology & Evolution, 34, 903–913. https://doi.org/10.1016/j.tree.2019.05.012 | Landscape connectivity alters the evolution of density-dependent dispersal during pushed range expansions | Maxime Dahirel, Aline Bertin, Vincent Calcagno, Camille Duraj, Simon Fellous, Géraldine Groussier, Eric Lombaert, Ludovic Mailleret, Anaël Marchand, Elodie Vercken | <p style="text-align: justify;">As human influence reshapes communities worldwide, many species expand or shift their ranges as a result, with extensive consequences across levels of biological organization. Range expansions can be ranked on a con... | | Evolutionary Ecology, Experimental Evolution | Inês Fragata | 2021-03-05 17:15:46 | ||
19 Jul 2021

Host phenology can drive the evolution of intermediate virulence strategies in some obligate-killer parasitesModelling parasitoid virulence evolution with seasonalityRecommended by Samuel Alizon based on reviews by Alex Best and 2 anonymous reviewers
The harm most parasites cause to their host, i.e. the virulence, is a mystery because host death often means the end of the infectious period. For obligate killer parasites, or “parasitoids”, that need to kill their host to transmit to other hosts the question is reversed. Indeed, more rapid host death means shorter generation intervals between two infections and mathematical models show that, in the simplest settings, natural selection should always favour more virulent strains (Levin and Lenski, 1983). Adding biological details to the model modifies this conclusion and, for instance, if the relationship between the infection duration and the number of parasites transmission stages produced in a host is non-linear, strains with intermediate levels of virulence can be favoured (Ebert and Weisser 1997). Other effects, such as spatial structure, could yield similar effects (Lion and van Baalen, 2007). In their study, MacDonald et al. (2021) explore another type of constraint, which is seasonality. Earlier studies, such as that by Donnelly et al. (2013) showed that this constraint can affect virulence evolution but they had focused on directly transmitted parasites. Using a mathematical model capturing the dynamics of a parasitoid, MacDonald et al. (2021) show if two main assumptions are met, namely that at the end of the season only transmission stages (or “propagules”) survive and that there is a constant decay of these propagules with time, then strains with intermediate levels of virulence are favoured. Practically, the authors use delay differential equations and an adaptive dynamics approach to identify evolutionary stable strategies. As expected, the longer the short the season length, the higher the virulence (because propagule decay matters less). The authors also identify a non-linear relationship between the variation in host development time and virulence. Generally, the larger the variation, the higher the virulence because the parasitoid has to kill its host before the end of the season. However, if the variation is too wide, some hosts become physically impossible to use for the parasite, whence a decrease in virulence. Finally, MacDonald et ali. (2021) show that the consequence of adding trade-offs between infection duration and the number of propagules produced is in line with earlier studies (Ebert and Weisser 1997). These mathematical modelling results provide testable predictions for using well-described systems in evolutionary ecology such as daphnia parasitoids, baculoviruses, or lytic phages. Reference Donnelly R, Best A, White A, Boots M (2013) Seasonality selects for more acutely virulent parasites when virulence is density dependent. Proc R Soc B, 280, 20122464. https://doi.org/10.1098/rspb.2012.2464 Ebert D, Weisser WW (1997) Optimal killing for obligate killers: the evolution of life histories and virulence of semelparous parasites. Proc R Soc B, 264, 985–991. https://doi.org/10.1098/rspb.1997.0136 Levin BR, Lenski RE (1983) Coevolution in bacteria and their viruses and plasmids. In: Futuyma DJ, Slatkin M eds. Coevolution. Sunderland, MA, USA: Sinauer Associates, Inc., 99–127. Lion S, van Baalen M (2008) Self-structuring in spatial evolutionary ecology. Ecol. Lett., 11, 277–295. https://doi.org/10.1111/j.1461-0248.2007.01132.x MacDonald H, Akçay E, Brisson D (2021) Host phenology can drive the evolution of intermediate virulence strategies in some obligate-killer parasites. bioRxiv, 2021.03.13.435259, ver. 8 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.03.13.435259 | Host phenology can drive the evolution of intermediate virulence strategies in some obligate-killer parasites | Hannelore MacDonald, Erol Akçay, Dustin Brisson | <p style="text-align: justify;">The traditional mechanistic trade-offs resulting in a negative correlation between transmission and virulence are the foundation of nearly all current theory on the evolution of parasite virulence. Several ecologica... | | Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Epidemiology, Evolutionary Theory | Samuel Alizon | 2021-03-14 13:47:33 | ||
08 Nov 2021

Dynamics of sex-biased gene expression over development in the stick insect Timema californicumSex-biased gene expression in an hemimetabolous insect: pattern during development, extent, functions involved, rate of sequence evolution, and comparison with an holometabolous insectRecommended by Nadia Aubin-Horth based on reviews by 2 anonymous reviewersAn individual’s sexual phenotype is determined during development. Understanding which pathways are activated or repressed during the developmental stages leading to a sexually mature individual, for example by studying gene expression and how its level is biased between sexes, allows us to understand the functional aspects of dimorphic phenotypes between the sexes. Several studies have quantified the differences in transcription between the sexes in mature individuals, showing the extent of this sex-bias and which functions are affected. There is, however, less data available on what occurs during the different phases of development leading to this phenotype, especially in species with specific developmental strategies, such as hemimetabolous insects. While many well-studied insects such as the honey bee, drosophila, and butterflies, exhibit an holometabolous development ("holo" meaning "complete" in reference to their drastic metamorphosis from the juvenile to the adult stage), hemimetabolous insects have juvenile stages that look similar to the adult stage (the hemi prefix meaning "half", referring to the more tissue-specific changes during development), as seen in crickets, cockroaches, and stick insects. Learning more about what happens during development in terms of the identity of genes that are sex-biased (are they the same genes at different developmental stages? What are their function? Do they exhibit specific sequence evolution rates? Is one sex over-represented in the sex-biased genes?) and their quantity over developmental time (gradual or abrupt increase in number, if any?) would allow us to better understand the evolution of sexual dimorphism at the gene expression level and how it relates to dimorphism at the organismic level. Djordjevic et al (2021) studied the transcriptome during development in an hemimetabolous stick insect, to improve our knowledge of this type of development, where the organismic phenotype is already mostly present in the early life stages. To do this, they quantified whole-genome gene expression levels in whole insects, using RNA-seq at three different developmental stages. One of the interesting results presented by Djordjevic and colleagues is that the increase in the number of genes that were sex-biased in expression is gradual over the three stages of development studied and it is mostly the same genes that stay sex-biased over time, reflecting the gradual change in phenotypes between hatchlings, juveniles and adults. Furthermore, male-biased genes had faster sequence divergence rates than unbiased genes and that female-biased genes. This new information of sex-bias in gene expression in an hemimetabolous insect allowed the authors to do a comparison of sex-biased genes with what has been found in a well-studied holometabolous insect, Drosophila. The gene expression patterns showed that four times more genes were sex-biased in expression in that species than in stick insects. Furthermore, the increase in the number of sex-biased genes during development was quite abrupt and clearly distinct in the adult stage, a pattern that was not seen in stick insects. As pointed out by the authors, this pattern of a "burst" of sex-biased genes at maturity is more common than the gradual increase seen in stick insects. With this study, we now know more about the evolution of sex-biased gene expression in an hemimetabolous insect and how it relates to their phenotypic dimorphism. Clearly, the next step will be to sample more hemimetabolous species at different life stages, to see how this pattern is widespread or not in this mode of development in insects. References Djordjevic J, Dumas Z, Robinson-Rechavi M, Schwander T, Parker DJ (2021) Dynamics of sex-biased gene expression during development in the stick insect Timema californicum. bioRxiv, 2021.01.23.427895, ver. 6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.01.23.427895 | Dynamics of sex-biased gene expression over development in the stick insect Timema californicum | Jelisaveta Djordjevic, Zoé Dumas, Marc Robinson-Rechavi, Tanja Schwander, Darren James Parker | <p style="text-align: justify;">Sexually dimorphic phenotypes are thought to arise primarily from sex-biased gene expression during development. Major changes in developmental strategies, such as the shift from hemimetabolous to holometabolous dev... | | Evo-Devo, Evolutionary Dynamics, Evolutionary Ecology, Expression Studies, Genotype-Phenotype, Molecular Evolution, Reproduction and Sex, Sexual Selection | Nadia Aubin-Horth | 2021-04-22 17:36:32 |




