
Latest recommendations

| Id | Title | Authors | Abstract | Picture | Thematic fields▲ | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
20 Dec 2022
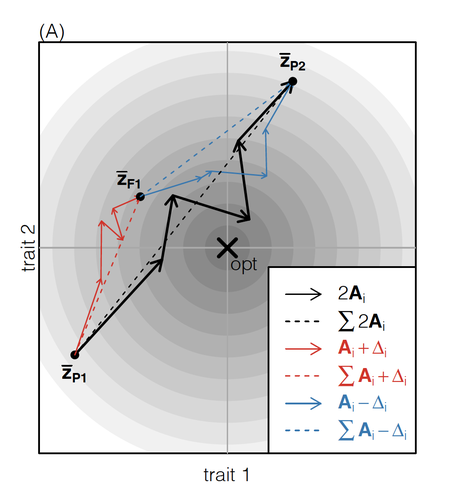
How does the mode of evolutionary divergence affect reproductive isolation?A general model of fitness effects following hybridisationRecommended by Matthew Hartfield based on reviews by Luis-Miguel Chevin and Juan LiStudying the effects of speciation, hybridisation, and evolutionary outcomes following reproduction from divergent populations is a major research area in evolutionary genetics [1]. There are two phenomena that have been the focus of contemporary research. First, a classic concept is the formation of ‘Bateson-Dobzhansky-Muller’ incompatibilities (BDMi) [2–4] that negatively affect hybrid fitness. Here, two diverging populations accumulate mutations over time that are unique to that subpopulation. If they subsequently meet, then these mutations might negatively interact, leading to a loss in fitness or even a complete lack of reproduction. BDMi formation can be complex, involving multiple genes and the fitness changes can depend on the direction of introgression [5]. Second, such secondary contact can instead lead to heterosis, where offspring are fitter than their parental progenitors [6]. Understanding which outcomes are likely to arise require one to know the potential fitness effects of mutations underlying reproductive isolation, to determine whether they are likely to reduce or enhance fitness when hybrids are formed. This is far from an easy task, as it requires one to track mutations at several loci, along with their effects, across a fitness landscape. The work of De Sanctis et al. [7] neatly fills in this knowledge gap, by creating a general mathematical framework for describing the consequences of a cross from two divergent populations. The derivations are based on Fisher’s Geometric Model, which is widely used to quantify selection acting on a general fitness landscape that is affected by several biological traits [8,9], and has previously been used in theoretical studies of hybridisation [10–12]. By doing so, they are able to decompose how divergence at multiple loci affects offspring fitness through both additive and dominance effects. A key result arising from their analyses is demonstrating how offspring fitness can be captured by two main functions. The first one is the ‘net effect of evolutionary change’ that, broadly defined, measures how phenotypically divergent two populations are. The second is the ‘total amount of evolutionary change’, which reflects how many mutations contribute to divergence and the effect sizes captured by each of them. The authors illustrate these measurements using simulations covering different scenarios, demonstrating how different parental states can lead to similar fitness outcomes. They also propose experimental methods to measure the underlying mutational effects. This study neatly demonstrates how complex genetic phenomena underlying hybridisation can be captured using fairly simple mathematical formulae. This powerful approach will thus open the door for future research to investigate hybridisation in more detail, whether it is by expanding on these theoretical models or using the elegant outcomes to quantify fitness effects in experiments.
References 1. Coyne JA, Orr HA. Speciation. Sunderland, Mass: Sinauer Associates; 2004. | How does the mode of evolutionary divergence affect reproductive isolation? | Bianca De Sanctis, Hilde Schneemann, John J. Welch | <p>When divergent populations interbreed, the outcome will be affected by the genomic and phenotypic differences that they have accumulated. In this way, the mode of evolutionary divergence between populations may have predictable consequences for... | | Adaptation, Evolutionary Theory, Hybridization / Introgression, Population Genetics / Genomics, Speciation | Matthew Hartfield | 2022-03-30 14:55:46 | ||
20 Dec 2016

POSTPRINT
Experimental Evolution of Gene Expression and Plasticity in Alternative Selective RegimesGenetic adaptation counters phenotypic plasticity in experimental evolutionRecommended by Luis-Miguel Chevin and Stephanie BedhommeHow do phenotypic plasticity and adaptive evolution interact in a novel or changing environment? Does evolution by natural selection generally reinforce initially plastic phenotypic responses, or does it instead oppose them? And to what extent does evolution of a trait involve evolution of its plasticity? These questions have lied at the heart of research on phenotypic evolution in heterogeneous environments ever since it was realized that the environment is likely to affect the expression of many (perhaps most) characters of an individual. Importantly, this broad definition of phenotypic plasticity as change in the average phenotype of a given genotype in response to its environment of development (or expression) does not involve any statement about the adaptiveness of the plastic changes. Theory on the evolution of plasticity has devoted much effort to understanding how reaction norm should evolve under different regimes of environmental change in space and time, and depending on genetic constraints on reaction norm shapes. However on an empirically ground, the questions above have mostly been addressed for individual traits, often chosen a priori for their likeliness to exhibit adaptive plasticity, and we still lack more systematic answers. These can be provided by so-called ‘phenomic’ approaches, where a large number of traits are tracked without prior information on their biological or ecological function. A problem is that the number of phenotypic characters that can be measured in an organism is virtually infinite (and to some extent arbitrary), and that scaling issues makes it difficult to compare different sets of traits. Gene-expression levels offer a partial solution to this dilemma, as they can be considered as a very large number of traits (one per typed gene) that can be measured easily and uniformly (fold change in the number of reads in RNAseq). As for any traits, expression levels of different genes may be genetically correlated, to an extent that depends on their regulation mechanism: cis-regulatory sequences that only affect expression of neighboring genes are likely to cause independent gene expression, while more systematic modifiers of expression (e.g. trans-regulators such as transcription factors) may cause correlated genetic responses of the expression of many genes. Huang and Agrawal [1] have studied plasticity and evolution of gene expression level in young larvae of populations of Drosophila melanogaster that have evolved for about 130 generations under either a constant environment (salt or cadmium), or an environment that is heterogeneous in time or space (combining salt and cadmium). They report a wealth of results, of which we summarize the most striking here. First, among genes that (i) were initially highly plastic and (ii) evolved significant divergence in expression levels between constant environment treatments, the evolved divergence is predominantly in the opposite direction to the initial plastic response. This suggests that either plasticity was initially maladaptive, or the selective pressure changed during the evolutionary process (see below). This somewhat unexpected result strikingly mirrors that from a study published last year in Nature [2], where the same pattern was found for responses of guppies to the presence of predators. However, Huang and Agrawal [1] went beyond this study by deciphering the underlying mechanisms in several interesting ways. First, they showed that change in gene expression often occurred at genes close to SNPs with differentiated frequencies across treatments (but not at genes with differentiated SNPs in their coding sequences), suggesting that cis-regulatory sequences are involved. This is also suggested by the fact that changes in gene expression are mostly caused by the increased expression of only one allele at polymorphic loci, and is a first step towards investigating the genetic underpinnings of (co)variation in gene expression levels. Another interesting set of findings concerns evolution of plasticity in treatments with variable environments. To compare the gene-expression plasticity that evolved in these treatments to an expectation, the authors considered that the expression levels in populations maintained for a long time under constant salt or cadmium had reached an optimum. The differences between these expression levels were thus assumed to predict the level of plasticity that should evolve in a heterogeneous environment (with both cadmium and salt) under perfect environmental predictability. The authors showed that plasticity did evolve more in the expected direction in heterogeneous than in constant environments, resulting in better adapted final expression levels across environments. Taken collectively, these results provide an unprecedented set of patterns that are greatly informative on how plasticity and evolution interact in constant versus changing environments. But of course, interpretations in terms of adaptive versus maladaptive plasticity are more challenging, as the authors themselves admit. Even though environmentally determined gene expression is the basic mechanism underlying the phenotypic plasticity of most traits, it is extremely difficult to relate to more integrated phenotypes for which we can understand the selection pressures, especially in multicellular organisms. The authors have recently investigated evolutionary change of quantitative traits in these selected lines, so it might be possible to establish links between reaction norms for macroscopic traits to those for gene expression levels. Such an approach would also involve tracking gene expression throughout life, rather than only in young larvae as done here, thus putting phenotypic complexity back in the picture also for expression levels. Another difficulty is that a plastic response that was originally adaptive may be replaced by an opposite evolutionary response in the long run, without having to invoke initially maladaptive plasticity. For instance, the authors mention the possibility that a generic stress response is initially triggered by cadmium, but is eventually unnecessary and costly after evolution of genetic mechanisms for cadmium detoxification (a case of so-called genetic accommodation). In any case, this study by Huang and Agrawal [1], together with the one by Ghalambor et al. last year [2], reports novel and unexpected results, which are likely to stimulate researchers interested in plasticity and evolution in heterogeneous environments for the years to come. References [1] Huang Y, Agrawal AF. 2016. Experimental Evolution of Gene Expression and Plasticity in Alternative Selective Regimes. PLoS Genetics 12:e1006336. doi: 10.1371/journal.pgen.1006336 [2] Ghalambor CK, Hoke KL, Ruell EW, Fischer EK, Reznick DN, Hughes KA. 2015. Non-adaptive plasticity potentiates rapid adaptive evolution of gene expression in nature. Nature 525: 372-375. doi: 10.1038/nature15256 | Experimental Evolution of Gene Expression and Plasticity in Alternative Selective Regimes | Huang Y, Agrawal AF | Little is known of how gene expression and its plasticity evolves as populations adapt to different environmental regimes. Expression is expected to evolve adaptively in all populations but only those populations experiencing environmental heterog... | | Adaptation, Experimental Evolution, Expression Studies, Phenotypic Plasticity | Luis-Miguel Chevin | 2016-12-20 09:04:15 | ||
03 Jun 2019

Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory networkEarly and late flowering gene expression patterns in maizeRecommended by Tanja Pyhäjärvi based on reviews by Laura Shannon and 2 anonymous reviewersArtificial selection experiments are key experiments in evolutionary biology. The demonstration that application of selective pressure across multiple generations results in heritable phenotypic changes is a tangible and reproducible proof of the evolution by natural selection. References [1] Hill, W. G., & Caballero, A. (1992). Artificial selection experiments. Annual Review of Ecology and Systematics, 23(1), 287-310. doi: 10.1146/annurev.es.23.110192.001443 | Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network | Maud Irène Tenaillon, Khawla Sedikki, Maeva Mollion, Martine Le Guilloux, Elodie Marchadier, Adrienne Ressayre, Christine Dillmann | <p>Artificial selection experiments are designed to investigate phenotypic evolution of complex traits and its genetic basis. Here we focused on flowering time, a trait of key importance for plant adaptation and life-cycle shifts. We undertook div... | | Adaptation, Experimental Evolution, Expression Studies, Quantitative Genetics | Tanja Pyhäjärvi | 2018-11-23 11:57:35 | ||
08 Aug 2018

Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutationsInbreeding compensates for reduced sexual selection in purging deleterious mutationsRecommended by Charles Baer based on reviews by 2 anonymous reviewersTwo evolutionary processes have been shown in theory to enhance the effects of natural selection in purging deleterious mutations from a population (here ""natural"" selection is defined as ""selection other than sexual selection""). First, inbreeding, especially self-fertilization, facilitates the removal of deleterious recessive alleles, the effects of which are largely hidden from selection in heterozygotes when mating is random. Second, sexual selection can facilitate the removal of deleterious alleles of arbitrary dominance, with little or no demographic cost, provided that deleterious effects are greater in males than in females (""genic capture""). Inbreeding (especially selfing) and sexual selection are often negatively correlated in nature. Empirical tests of the role of sexual selection in purging deleterious mutations have been inconsistent, potentially due to the positive relationship between sexual selection and intersexual genetic conflict. References [1] Noël, E., Fruitet, E., Lelaurin, D., Bonel, N., Segard, A., Sarda, V., Jarne, P., & David P. (2018). Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutations. bioRxiv, 273367, ver. 3 recommended and peer-reviewed by PCI Evol Biol. doi: 10.1101/273367 | Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutations | E. Noël, E. Fruitet, D. Lelaurin, N. Bonel, A. Ségard, V. Sarda, P. Jarne and P. David | <p>This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (https://dx.doi.org/10.24072/pci.evolbiol.100055). Theory and empirical data showed that two processes can boost selection against deleterious mutations, ... | | Adaptation, Experimental Evolution, Reproduction and Sex, Sexual Selection | Charles Baer | Anonymous | 2018-03-01 08:12:37 | |
24 Mar 2023
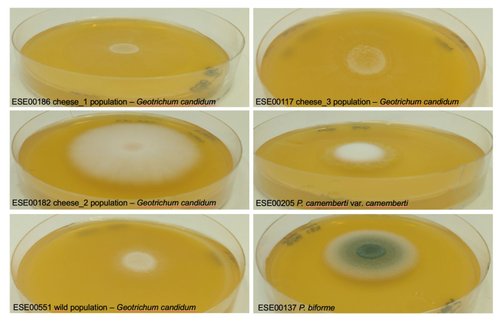
Domestication of different varieties in the cheese-making fungus Geotrichum candidumDiverse outcomes in cheese fungi domesticationRecommended by Christelle Fraïsse based on reviews by Delphine Sicard and 1 anonymous reviewer based on reviews by Delphine Sicard and 1 anonymous reviewer
Domestication is a complex process that imprints the demography and the genomes of domesticated populations, enforcing strong selective pressures on traits favourable to humans, e.g. for food production [1]. Domestication has been quite intensely studied in plants and animals, but less so in micro-organisms such as fungi, despite their assets (e.g. their small genomes and tractability in the lab). This elegant study by Bennetot and collaborators [2] on the cheese-making fungus Geotrichum candidum adds to the mounting body of studies in the genomics of fungi, proving they are excellent models in evolutionary biology for studying adaptation and drift in eukaryotes [3]. Bennetot et al. newly showed with whole genome sequences that all G. candidum strains isolated from cheese form a monophyletic clade subdivided into three genetically differentiated populations with several admixed strains, while the wild strains sampled from diverse geographic locations form a sister clade. This suggests the wild progenitor was not sampled in the present study and calls for future exciting work on the domestication history of the G. candidum fungus. The authors scanned the genomes for footprints of adaptation to the cheese environment and identified promising candidates, such as a gene involved in iron uptake (this element is limiting in cheese). Their functional genome analysis also provides evidence for higher contents of transposable elements in cheese-making strains, likely due to relaxed selection during the domestication process. This paper is particularly impressive in that the authors complemented the population genomic approach with the phenotypic characterization of the strains and tested their ability to outcompete common fungal food spoilers. The authors convincingly showed that cheese-making strains display phenotypic differences relative to wild relatives for multiple traits such as slower growth, lower proteolysis activity and a greater amount of volatiles attractive to consumers, these phenotypes being beneficial for cheese making. Finally, this work is particularly inspiring because it thoroughly discusses convergent evolution during domestication in different cheese-associated fungi. Indeed, studying populations experiencing similar environmental pressures is fundamental to understanding whether evolution is repeatable [4]. For instance, all three cheese populations of G. candidum exhibit a lower genetic diversity than wild populations. However, only one population displays a stronger domestication syndrome, resembling the Penicillium camemberti situation [5]. Furthermore, different cheese-making practices may have led to varying situations with clonal lineages in non-Roquefort P. roqueforti and P. camemberti [5, 6], while the cheese-making G. candidum populations still harbour some diversity. In a nutshell, Bennetot's study makes an important contribution to evolutionary biology and highlights the value of diversifying our model organisms toward under-represented clades. REFERENCES [1] Diamond J (2002) Evolution, consequences and future of plant and animal domestication. Nature 418: 700–707. https://doi.org/10.1038/nature01019 [2] Bennetot B, Vernadet J-P, Perkins V, Hautefeuille S, Rodríguez de la Vega RC, O’Donnell S, Snirc A, Grondin C, Lessard M-H, Peron A-C, Labrie S, Landaud S, Giraud T, Ropars J (2023) Domestication of different varieties in the cheese-making fungus Geotrichum candidum. bioRxiv, 2022.05.17.492043, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.05.17.492043 [3] Gladieux P, Ropars J, Badouin H, Branca A, Aguileta G, de Vienne DM, Rodríguez de la Vega RC, Branco S, Giraud T (2014) Fungal evolutionary genomics provides insight into the mechanisms of adaptive divergence in eukaryotes. Mol. Ecol. 23: 753–773. https://doi.org/10.1111/mec.12631 [4] Bolnick DI, Barrett RD, Oke KB, Rennison DJ, Stuart YE (2018) (Non)Parallel evolution. Ann. Rev. Ecol. Evol. Syst. 49: 303–330. https://doi.org/10.1146/annurev-ecolsys-110617-062240 [5] Ropars J, Didiot E, Rodríguez de la Vega RC, Bennetot B, Coton M, Poirier E, Coton E, Snirc A, Le Prieur S, Giraud T (2020) Domestication of the Emblematic White Cheese-Making Fungus Penicillium camemberti and Its Diversification into Two Varieties. Current Biol. 30: 4441–4453.e4. https://doi.org/10.1016/j.cub.2020.08.082 [6] Dumas, E, Feurtey, A, Rodríguez de la Vega, RC, Le Prieur S, Snirc A, Coton M, Thierry A, Coton E, Le Piver M, Roueyre D, Ropars J, Branca A, Giraud T (2020) Independent domestication events in the blue-cheese fungus Penicillium roqueforti. Mol Ecol. 29: 2639–2660. https://doi.org/10.1111/mec.15359 | Domestication of different varieties in the cheese-making fungus *Geotrichum candidum* | Bastien Bennetot, Jean-Philippe Vernadet, Vincent Perkins, Sophie Hautefeuille, Ricardo C. Rodríguez de la Vega, Samuel O’Donnell, Alodie Snirc, Cécile Grondin, Marie-Hélène Lessard, Anne-Claire Peron, Steve Labrie, Sophie Landaud, Tatiana Giraud,... | <p>Domestication is an excellent model for studying adaptation processes, involving recent adaptation and diversification, convergence following adaptation to similar conditions, as well as degeneration of unused functions. <em>Geotrichum candidum... | | Adaptation, Genome Evolution, Population Genetics / Genomics | Christelle Fraïsse | 2022-08-12 20:50:42 | ||
13 Dec 2016

POSTPRINT
A supergene determines highly divergent male reproductive morphs in the ruffSupergene Control of a Reproductive PolymorphismRecommended by Thomas Flatt and Laurent KellerTwo back-to-back papers published earlier this year in Nature Genetics provide compelling evidence for the control of a male reproductive polymorphism in a wading bird by a "supergene", a cluster of tightly linked genes [1-2]. The bird in question, the ruff (Philomachus pugnax), has a rather unusual reproductive system that consists of three distinct types of males ("reproductive morphs"): aggressive "independents" who represent the majority of males; a smaller fraction of non-territorial "satellites" who are submissive towards "independents"; and "faeders" who mimic females and are rare. Previous work has shown that the male morphs differ in major aspects of mating and aggression behavior, plumage coloration and body size, and that – intriguingly – this complex multi-trait polymorphism is apparently controlled by a single autosomal Mendelian locus with three alleles [3]. To uncover the genetic control of this polymorphism two independent teams, led by Terry Burke [1] and Leif Andersson [2], have set out to analyze the genomes of male ruffs. Using a combination of genomics and genetics, both groups managed to pin down the supergene locus and map it to a non-recombining, 4.5 Mb large inversion which arose 3.8 million years ago. While "independents" are homozygous for the ancestral uninverted sequence, "satellites" and "faeders" carry evolutionarily divergent, dominant alternative haplotypes of the inversion. Thus, as in several other notable cases, for example the supergene control of disassortative mating, aggressiveness and plumage color in white-throated sparrows [4], of mimicry in Heliconius and Papilio butterflies [5-6], or of social structure in ants [7], an inversion – behaving as a single "locus" – underpins the mechanistic basis of the supergene. More generally, and beyond inversions, a growing number of studies now shows that selection can favor the evolution of suppressed recombination, thereby leading to the emergence of clusters of tightly linked loci which can then control – presumably due to polygenic gene action – a suite of complex phenotypes [8-10]. A largely unresolved question in this field concerns the identity of the causative alleles and loci within a given supergene. Recent progress on this question has been made for example in Papilio polytes butterflies where a mimicry supergene has been found to involve – surprisingly – only a single but large gene: multiple mimicry alleles in the doublesex gene are maintained in strong linkage disequilibrium via an inversion. It will clearly be of great interest to see future examples of such a fine-scale genetic dissection of supergenes. In conclusion, we were impressed by the data and analyses of Küpper et al. [1] and Lamichhaney et al. [2]: both papers beautifully illustrate how genomics and evolutionary ecology can be combined to make new, exciting discoveries. Both papers will appeal to readers with an interest in supergenes, inversions, the interplay of selection and recombination, or the genetic control of complex phenotypes. References [1] Küpper C, Stocks M, Risse JE, dos Remedios N, Farrell LL, McRae SB, Morgan TC, Karlionova N, Pinchuk P, Verkuil YI, et al. 2016. A supergene determines highly divergent male reproductive morphs in the ruff. Nature Genetics 48:79-83. doi: 10.1038/ng.3443 [2] Lamichhaney S, Fan G, Widemo F, Gunnarsson U, Thalmann DS, Hoeppner MP, Kerje S, Gustafson U, Shi C, Zhang H, et al. 2016. Structural genomic changes underlie alternative reproductive strategies in the ruff (Philomachus pugnax). Nature Genetics 48:84-88. doi: 10.1038/ng.3430 [3] Lank DB, Smith CM, Hanotte O, Burke T, Cooke F. 1995. Genetic polymorphism for alternative mating behaviour in lekking male ruff Philomachus pugnax. Nature 378:59-62. doi: 10.1038/378059a0 [4] Tuttle Elaina M, Bergland Alan O, Korody Marisa L, Brewer Michael S, Newhouse Daniel J, Minx P, Stager M, Betuel A, Cheviron Zachary A, Warren Wesley C, et al. 2016. Divergence and Functional Degradation of a Sex Chromosome-like Supergene. Current Biology 26:344-350. doi: 10.1016/j.cub.2015.11.069 [5] Joron M, Frezal L, Jones RT, Chamberlain NL, Lee SF, Haag CR, Whibley A, Becuwe M, Baxter SW, Ferguson L, et al. 2011. Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 477:203-206. doi: 10.1038/nature10341 [6] Kunte K, Zhang W, Tenger-Trolander A, Palmer DH, Martin A, Reed RD, Mullen SP, Kronforst MR. 2014. doublesex is a mimicry supergene. Nature 507:229-232. doi: 10.1038/nature13112 [7] Wang J, Wurm Y, Nipitwattanaphon M, Riba-Grognuz O, Huang Y-C, Shoemaker D, Keller L. 2013. A Y-like social chromosome causes alternative colony organization in fire ants. Nature 493:664-668. doi: 10.1038/nature11832 [8] Thompson MJ, Jiggins CD. 2014. Supergenes and their role in evolution. Heredity 113:1-8. doi: 10.1038/hdy.2014.20 [9] Schwander T, Libbrecht R, Keller L. 2014. Supergenes and Complex Phenotypes. Current Biology 24:R288-R294. doi: 10.1016/j.cub.2014.01.056 [10] Charlesworth D. 2015. The status of supergenes in the 21st century: recombination suppression in Batesian mimicry and sex chromosomes and other complex adaptations. Evolutionary Applications 9:74-90. doi: 10.1111/eva.12291 | A supergene determines highly divergent male reproductive morphs in the ruff | Küpper C, Stocks M, Risse JE, dos Remedios N, Farrell LL, McRae SB, Morgan TC, Karlionova N, Pinchuk P, Verkuil YI, et al. | Three strikingly different alternative male mating morphs (aggressive 'independents', semicooperative 'satellites' and female-mimic 'faeders') coexist as a balanced polymorphism in the ruff, *Philomachus pugnax*, a lek-breeding wading bird1, 2, 3.... | | Adaptation, Genotype-Phenotype, Life History, Population Genetics / Genomics, Reproduction and Sex | Thomas Flatt | 2016-12-13 17:28:13 | ||
02 Sep 2022

Introgression between highly divergent sea squirt genomes: an adaptive breakthrough?A match made in the Anthropocene: human-mediated adaptive introgression across a speciation continuumRecommended by Fernando Racimo based on reviews by Michael Westbury, Andrew Foote and Erin CalfeeThe long-distance transport and introduction of new species by humans is increasingly leading divergent lineages to interact, and sometimes interbreed, even after thousands or millions of years of separation. It is thus of prime importance to understand the consequences of these contemporary admixture events on the evolutionary fitness of interacting organisms, and their ecological implications. Ciona robusta and Ciona intestinalis are two species of sea squirts that diverged between 1.5 and 2 million years ago and recently came into contact again. This occurred through human-mediated introduction of C. robusta (native to the Northwest Pacific) into the range of C. intestinalis (the English channeled Northeast Atlantic). In this study, Fraïsse et al. (2022) follow up on earlier work by Le Moan et al. (2021), who had identified a long genomic hotspot of introgression of C. robusta ancestry segments in chromosome 5 of C. intestinalis. The hotspot bears suggestive evidence of positive selection and the authors aimed to investigate this further using fully phased whole-genome sequences. The authors narrow down on the exact boundaries of the introgressed region, and make a compelling case that it has been the likely target of positive selection after introgression, using various complementary approaches based on patterns of population differentiation, haplotype structure and local levels of diversity in the region. Using extensive demographic modeling, they also show that the introgression event was likely recent (approximately 75 years ago), and distinct from other tracts in the C. intestinalis genome that are likely a product of more ancient episodes of interbreeding in the past 30,000 years. Narrowing down on the potential drivers of selection, the authors show that candidate SNPs in the region overlap with the cytochrome family 2 subfamily U gene - involved in the detoxification of exogenous compounds - potentially reflecting adaptation to chemicals encountered in the sea squirt's environment. There also appears to be copy number variation at the candidate SNPs, which provides clues into the adaptation mechanism in the region. All reviewers agreed that the work carried out by the authors is elegant and the results are robustly supported and well presented. In a round of reviews, various clarifications of the manuscript were suggested by the reviewers, including on the quality of the newly generated sequencing data, and some suggestions for qualifications on the conclusions reached by the authors as well as changes in the figures to increase their clarity. The authors addressed the different concerns of the reviewers, and the new version is much improved. This study into human-mediated introgression and its consequences for adaptation is, in my view, both well thought-out and executed. I therefore provide an enthusiastic recommendation of this manuscript. References Fraïsse C, Le Moan A, Roux C, Dubois G, Daguin-Thiébaut C, Gagnaire P-A, Viard F and Bierne N (2022) Introgression between highly divergent sea squirt genomes: an adaptive breakthrough? bioRxiv, 2022.03.22.485319, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.03.22.485319 Le Moan A, Roby C, Fraïsse C, Daguin-Thiébaut C, Bierne N, Viard F (2021) An introgression breakthrough left by an anthropogenic contact between two ascidians. Molecular Ecology, 30, 6718–6732. https://doi.org/10.1111/mec.16189 | Introgression between highly divergent sea squirt genomes: an adaptive breakthrough? | Christelle Fraïsse, Alan Le Moan, Camille Roux, Guillaume Dubois, Claire Daguin-Thiébaut, Pierre-Alexandre Gagnaire, Frédérique Viard, Nicolas Bierne | <p style="text-align: justify;">Human-mediated introductions are reshuffling species distribution on a global scale. Consequently, an increasing number of allopatric taxa are now brought into contact, promoting introgressive hybridization between ... | | Adaptation, Hybridization / Introgression, Population Genetics / Genomics | Fernando Racimo | 2022-04-14 15:30:42 | ||
03 Apr 2020

Evolution at two time-frames: ancient and common origin of two structural variants involved in local adaptation of the European plaice (Pleuronectes platessa)Genomic structural variants involved in local adaptation of the European plaiceRecommended by Maren Wellenreuther based on reviews by 3 anonymous reviewersAwareness has been growing that structural variants in the genome of species play a fundamental role in adaptive evolution and diversification [1]. Here, Le Moan and co-authors [2] report empirical genomic-wide SNP data on the European plaice (Pleuronectes platessa) across a major environmental transmission zone, ranging from the North Sea to the Baltic Sea. Regions of high linkage disequilibrium suggest the presence of two structural variants that appear to have evolved 220 kya. These two putative structural variants show weak signatures of isolation by distance when contrasted against the rest of the genome, but the frequency of the different putative structural variants appears to co-vary in some parts of the studied range with the environment, indicating the involvement of both selective and neutral processes. This study adds to the mounting body of evidence that structural genomic variants harbour significant information that allows species to respond and adapt to the local environmental context. References [1] Wellenreuther, M., Mérot, C., Berdan, E., & Bernatchez, L. (2019). Going beyond SNPs: the role of structural genomic variants in adaptive evolution and species diversification. Molecular ecology, 28(6), 1203-1209. doi: 10.1111/mec.15066 | Evolution at two time-frames: ancient and common origin of two structural variants involved in local adaptation of the European plaice (Pleuronectes platessa) | Alan Le Moan, Dorte Bekkevold & Jakob Hemmer-Hansen | <p>Changing environmental conditions can lead to population diversification through differential selection on standing genetic variation. Structural variant (SV) polymorphisms provide examples of ancient alleles that in time become associated with... | | Adaptation, Hybridization / Introgression, Population Genetics / Genomics, Speciation | Maren Wellenreuther | 2019-07-13 12:44:01 | ||
02 Nov 2022
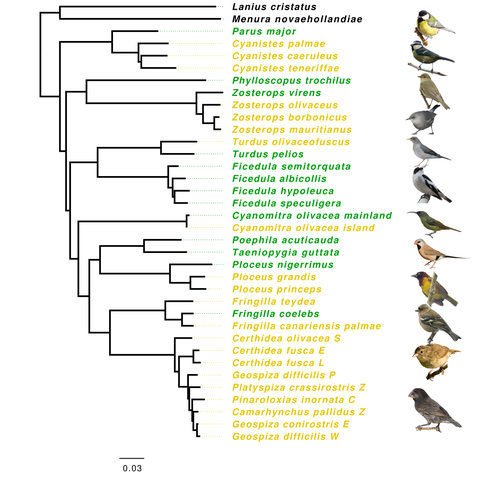
Evolution of immune genes in island birds: reduction in population sizes can explain island syndromeDemographic effects may affect adaptation to islandsRecommended by Emma Berdan based on reviews by Steven Fiddaman and 3 anonymous reviewersThe unique challenges associated with living on an island often result in organisms displaying a specific suite of traits commonly referred to as “island syndrome” (Adler and Levins, 1994; Burns, 2019; Baeckens and Van Damme, 2020). Large phenotypic shifts such as changes in size (e.g. shifts to gigantism or dwarfism, Lomolino, 2005) or coloration (Doutrelant et al., 2016) abound in the literature. However, less obvious phenotypes may also play a key role in adaptation to islands. One such trait, reduced immune function, has important implications for the future of island populations in the face of anthropogenic-induced changes. Due to lower parasite pressure caused by a less diverse and less virulent parasite population, island hosts may show a decrease in immune defenses (Beadell et al., 2006; Pérez‐Rodríguez et al., 2013). However, this hypothesis has been challenged, as many studies have found ambiguous or conflicting results (Matson, 2006; Illera et al., 2015). While most previous work has examined various immunological parameters (e.g., antibody concentrations), here, Barthe et al. (2022) take the novel approach of examining molecular signatures of immune genes. Using comparative genomic data from 34 different species of birds the authors examine the ratio of synonymous substitutions (i.e., not changing an amino acid) to non-synonymous substitutions (i.e., changing an amino acid) in innate and acquired immune genes (Pn/Ps ratio). Because population sizes on islands are lower which will affect molecular evolution, they compare these results to data from 97 control genes. Assuming relaxed selection on islands predicts that the difference between the Pn/Ps ratio of immune genes and of control genes (ΔPn/Ps) is greater in island species compared to mainland ones. As with previous work the authors found that the results differ depending on the category of immune genes. Both forms of innate defense: beta-defensins and Toll-like receptors did not show higher ΔPn/Ps for island populations. As these genes still have a higher Pn/Ps than control genes, the authors argue these results are in line with these genes being under purifying selection but lacking an “island effect”. Instead, the authors argue that demographic effects (i.e., reductions in Ne) may lead to the decreased immunity documented in other studies. In contrast, there was a reduction in Pn/Ps in MHC II genes, known to be under balancing selection. This reduction was stronger in island species and thus the authors argue that this is the only class of genes where a role for relaxed selection can be invoked. Together these results demonstrate that the changes in immunity experienced by island species are complex and that different categories of immune genes can experience different selective pressures. By including control genes in their study, they particularly highlight the importance of accounting for shifts in Ne when examining patterns of island species evolution. Hopefully, this kind of framework will be applied to other taxa to determine if these results are widespread or more specific to birds. References Adler GH, Levins R (1994) The Island Syndrome in Rodent Populations. The Quarterly Review of Biology, 69, 473–490. https://doi.org/10.1086/418744 Baeckens S, Van Damme R (2020) The island syndrome. Current Biology, 30, R338–R339. https://doi.org/10.1016/j.cub.2020.03.029 Barthe M, Doutrelant C, Covas R, Melo M, Illera JC, Tilak M-K, Colombier C, Leroy T, Loiseau C, Nabholz B (2022) Evolution of immune genes in island birds: reduction in population sizes can explain island syndrome. bioRxiv, 2021.11.21.469450, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.11.21.469450 Beadell JS, Ishtiaq F, Covas R, Melo M, Warren BH, Atkinson CT, Bensch S, Graves GR, Jhala YV, Peirce MA, Rahmani AR, Fonseca DM, Fleischer RC (2006) Global phylogeographic limits of Hawaii’s avian malaria. Proceedings of the Royal Society B: Biological Sciences, 273, 2935–2944. https://doi.org/10.1098/rspb.2006.3671 Burns KC (2019) Evolution in Isolation: The Search for an Island Syndrome in Plants. Cambridge University Press, Cambridge. https://doi.org/10.1017/9781108379953 Doutrelant C, Paquet M, Renoult JP, Grégoire A, Crochet P-A, Covas R (2016) Worldwide patterns of bird colouration on islands. Ecology Letters, 19, 537–545. https://doi.org/10.1111/ele.12588 Illera JC, Fernández-Álvarez Á, Hernández-Flores CN, Foronda P (2015) Unforeseen biogeographical patterns in a multiple parasite system in Macaronesia. Journal of Biogeography, 42, 1858–1870. https://doi.org/10.1111/jbi.12548 Lomolino MV (2005) Body size evolution in insular vertebrates: generality of the island rule. Journal of Biogeography, 32, 1683–1699. https://doi.org/10.1111/j.1365-2699.2005.01314.x Matson KD (2006) Are there differences in immune function between continental and insular birds? Proceedings of the Royal Society B: Biological Sciences, 273, 2267–2274. https://doi.org/10.1098/rspb.2006.3590 Pérez-Rodríguez A, Ramírez Á, Richardson DS, Pérez-Tris J (2013) Evolution of parasite island syndromes without long-term host population isolation: parasite dynamics in Macaronesian blackcaps Sylvia atricapilla. Global Ecology and Biogeography, 22, 1272–1281. https://doi.org/10.1111/geb.12084 | Evolution of immune genes in island birds: reduction in population sizes can explain island syndrome | Mathilde BARTHE, Claire DOUTRELANT, Rita COVAS, Martim MELO, Juan Carlos ILLERA, Marie-Ka TILAK, Constance COLOMBIER, Thibault LEROY , Claire LOISEAU , Benoit NABHOLZ | <p style="text-align: justify;">Shared ecological conditions encountered by species that colonize islands often lead to the evolution of convergent phenotypes, commonly referred to as “island syndrome”. Reduced immune functions have been previousl... | | Adaptation, Molecular Evolution, Population Genetics / Genomics | Emma Berdan | 2021-11-28 11:01:31 | ||
13 Dec 2018
Separate the wheat from the chaff: genomic analysis of local adaptation in the red coral Corallium rubrumPros and Cons of local adaptation scansRecommended by Guillaume Achaz based on reviews by Lucas Gonçalves da Silva and 1 anonymous reviewerThe preprint by Pratlong et al. [1] is a well thought quest for genomic regions involved in local adaptation to depth in a species a red coral living the Mediterranean Sea. It first describes a pattern of structuration and then attempts to find candidate genes involved in local adaptation by contrasting deep with shallow populations. Although the pattern of structuration is clear and meaningful, the candidate genomic regions involved in local adaptation remain to be confirmed. Two external reviewers and myself found this preprint particularly interesting regarding the right-mindedness of the authors in front of the difficulties they encounter during their experiments. The discussions on the pros and cons of the approach are very sound and can be easily exported to a large number of studies that hunt for local adaptation. In this sense, the lessons one can learn by reading this well documented manuscript are certainly valuable for a wide range of evolutionary biologists. References [1] Pratlong, M., Haguenauer, A., Brener, K., Mitta, G., Toulza, E., Garrabou, J., Bensoussan, N., Pontarotti P., & Aurelle, D. (2018). Separate the wheat from the chaff: genomic scan for local adaptation in the red coral Corallium rubrum. bioRxiv, 306456, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/306456 | Separate the wheat from the chaff: genomic analysis of local adaptation in the red coral Corallium rubrum | Pratlong M, Haguenauer A, Brener K, Mitta G, Toulza E, Garrabou J, Bensoussan N, Pontarotti P, Aurelle D | <p>Genomic data allow an in-depth and renewed study of local adaptation. The red coral (Corallium rubrum, Cnidaria) is a highly genetically structured species and a promising model for the study of adaptive processes along an environmental gradien... | | Adaptation, Population Genetics / Genomics | Guillaume Achaz | 2018-04-24 11:27:40 |




