
Latest recommendations

| Id | Title▲ | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
17 Feb 2020
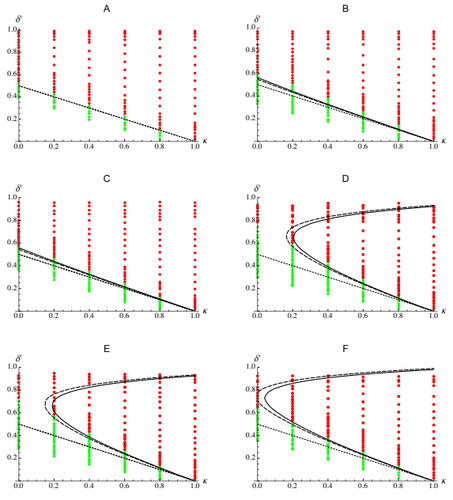
Epistasis, inbreeding depression and the evolution of self-fertilizationEpistasis and the evolution of selfingRecommended by Sylvain Gandon based on reviews by Nick Barton and 1 anonymous reviewerThe evolution of selfing results from a balance between multiple evolutionary forces. Selfing provides an "automatic advantage" due to the higher efficiency of selfers to transmit their genes via selfed and outcrossed offspring. Selfed offspring, however, may suffer from inbreeding depression. In principle the ultimate evolutionary outcome is easy to predict from the relative magnitude of these two evolutionary forces [1,2]. Yet, several studies explicitly taking into account the genetic architecture of inbreeding depression noted that these predictions are often too restrictive because selfing can evolve in a broader range of conditions [3,4]. References [1] Holsinger, K. E., Feldman, M. W., and Christiansen, F. B. (1984). The evolution of self-fertilization in plants: a population genetic model. The American Naturalist, 124(3), 446-453. doi: 10.1086/284287 | Epistasis, inbreeding depression and the evolution of self-fertilization | Diala Abu Awad and Denis Roze | <p>Inbreeding depression resulting from partially recessive deleterious alleles is thought to be the main genetic factor preventing self-fertilizing mutants from spreading in outcrossing hermaphroditic populations. However, deleterious alleles may... | | Evolutionary Theory, Quantitative Genetics, Reproduction and Sex | Sylvain Gandon | 2019-10-18 09:29:41 | ||
28 Sep 2020

Evolution and genetic architecture of disassortative mating at a locus under heterozygote advantageEvolutionary insights into disassortative mating and its association to an ecologically relevant supergeneRecommended by Charles Mullon based on reviews by Tom Van Dooren and 2 anonymous reviewers based on reviews by Tom Van Dooren and 2 anonymous reviewers
Heliconius butterflies are famous for their colorful wing patterns acting as a warning of their chemical defenses [1]. Most species are involved in Müllerian mimicry assemblies, as predators learn to associate common wing patterns with unpalatability and preferentially target rare variants. Such positive-frequency dependent selection homogenizes wing patterns at different localities, and in several species, all individuals within a community belong to the same morph [2]. In this respect, H. numata stands out. This species shows stable local polymorphism across multiple localities, with local populations home to up to seven distinct morphs [2]. Although a balance between migration and local positive-frequency dependent selection can allow some degree of local polymorphism, theory suggests that this occurs only when migration is within a narrow window [3]. References [1] Merrill, R M, K K Dasmahapatra, J W Davey, D D Dell'Aglio, J J Hanly, B Huber, C D Jiggins, et al. (2015). The Diversification of Heliconius butterflies: What Have We Learned in 150 Years? Journal of Evolutionary Biology 28 (8), 1417–38. https://doi.org/10.1111/jeb.12672. | Evolution and genetic architecture of disassortative mating at a locus under heterozygote advantage | Ludovic Maisonneuve, Mathieu Joron, Mathieu Chouteau and Violaine Llaurens | <p>The evolution of mate preferences may depend on natural selection acting on the mating cues and on the underlying genetic architecture. While the evolution of assortative mating with respect to locally adapted traits has been well-characterized... | | Evolutionary Theory, Population Genetics / Genomics, Reproduction and Sex, Sexual Selection | Charles Mullon | 2019-10-29 09:55:18 | ||
12 Jun 2017

Evolution and manipulation of vector host choiceModelling the evolution of how vector-borne parasites manipulate the vector's host choiceRecommended by Samuel Alizon based on reviews by Samuel Alizon and Nicole Mideo
Many parasites can manipulate their hosts, thus increasing their transmission to new hosts [1]. This is particularly the case for vector-borne parasites, which can alter the feeding behaviour of their hosts. However, predicting the optimal strategy is not straightforward because three actors are involved and the interests of the parasite may conflict with that of the vector. There are few models that consider the evolution of host manipulation by parasites [but see 2-4], but there are virtually none that investigated how parasites can manipulate the host choice of vectors. Even on the empirical side, many aspects of this choice remain unknown. Gandon [5] develops a simple evolutionary epidemiology model that allows him to formulate clear and testable predictions. These depend on which actor controls the trait (the vector or the parasite) and, when there is manipulation, whether it is realised via infected hosts (to attract vectors) or infected vectors (to change host choice). In addition to clarifying the big picture, Gandon [5] identifies some nice properties of the model, for instance an independence of the density/frequency-dependent transmission assumption or a backward bifurcation at R0=1, which suggests that parasites could persist even if their R0 is driven below unity. Overall, this study calls for further investigation of the different scenarios with more detailed models and experimental validation of general predictions. References [1] Hughes D, Brodeur J, Thomas F. 2012. Host manipulation by parasites. Oxford University Press. [2] Brown SP. 1999. Cooperation and conflict in host-manipulating parasites. Proceedings of the Royal Society of London B: Biological Sciences 266: 1899–1904. doi: 10.1098/rspb.1999.0864 [3] Lion S, van Baalen M, Wilson WG. 2006. The evolution of parasite manipulation of host dispersal. Proceedings of the Royal Society of London B: Biological Sciences. 273: 1063–1071. doi: 10.1098/rspb.2005.3412 [4] Vickery WL, Poulin R. 2010. The evolution of host manipulation by parasites: a game theory analysis. Evolutionary Ecology 24: 773–788. doi: 10.1007/s10682-009-9334-0 [5] Gandon S. 2017. Evolution and manipulation of vector host choice. bioRxiv 110577, ver. 3 of 7th June 2017. doi: 10.1101/110577 | Evolution and manipulation of vector host choice | Sylvain Gandon | The transmission of many animal and plant diseases relies on the behavior of arthropod vectors. In particular, the choice to feed on either infected or uninfected hosts can dramatically affect the epidemiology of vector-borne diseases. I develop a... | | Evolutionary Ecology, Evolutionary Epidemiology, Evolutionary Theory | Samuel Alizon | 2017-03-03 19:18:54 | ||
06 Oct 2017

Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebratesCombining molecular information on chromatin organisation with eQTLs and evolutionary conservation provides strong candidates for the evolution of gene regulation in mammalian brainsRecommended by Marc Robinson-Rechavi based on reviews by Marc Robinson-Rechavi and Charles DankoIn this manuscript [1], Francisco J. Novo proposes candidate non-coding genomic elements regulating neurodevelopmental genes. What is very nice about this study is the way in which public molecular data, including physical interaction data, is used to leverage recent advances in our understanding to molecular mechanisms of gene regulation in an evolutionary context. More specifically, evolutionarily conserved non coding sequences are combined with enhancers from the FANTOM5 project, DNAse hypersensitive sites, chromatin segmentation, ChIP-seq of transcription factors and of p300, gene expression and eQTLs from GTEx, and physical interactions from several Hi-C datasets. The candidate regulatory regions thus identified are linked to candidate regulated genes, and the author shows their potential implication in brain development. While the results are focused on a small number of genes, this allows to verify features of these candidates in great detail. This study shows how functional genomics is increasingly allowing us to fulfill the promises of Evo-Devo: understanding the molecular mechanisms of conservation and differences in morphology. References [1] Novo, FJ. 2017. Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebrates. bioRxiv, 150482, ver. 4 of Sept 29th, 2017. doi: 10.1101/150482 | Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebrates | Francisco J. Novo | <p>Many non-coding regulatory elements conserved in vertebrates regulate the expression of genes involved in development and play an important role in the evolution of morphology through the rewiring of developmental gene networks. Available biolo... | | Genome Evolution | Marc Robinson-Rechavi | Marc Robinson-Rechavi, Charles Danko | 2017-06-29 08:55:41 | |
24 Oct 2022

Evolutionary responses of energy metabolism, development, and reproduction to artificial selection for increasing heat tolerance in Drosophila subobscuraThe other side of the evolution of heat tolerance: correlated responses in metabolism and life-history traitsRecommended by Inês Fragata and Pedro Simões based on reviews by Marija Savić Veselinović and 1 anonymous reviewer
Understanding how species respond to environmental changes is becoming increasingly important in order to predict the future of biodiversity and species distributions under current global warming conditions (Rezende 2020; Bennett et al 2021). Two key factors to take into account in these predictions are the tolerance of organisms to heat stress and subsequently how they adapt to increasingly warmer temperatures. Coupled with this, one important factor that is often overlooked when addressing the evolution of thermal tolerance, is the correlated responses in traits that are important to fitness, such as life histories, behavior and the underlying metabolic processes. The rate and intensity of the thermal stress are expected to be major factors in shaping the evolution of heat tolerance and correlated responses in other traits. For instance, lower rates of thermal stress are predicted to select for individuals with a slower metabolism (Santos et al 2012), whereas low metabolism is expected to lead to a lower reproductive rate (Dammhahn et al 2018). To quantify the importance of the rate and intensity of thermal stress on the evolutionary response of heat tolerance and correlated response in behavior, Mesas et al (2021) performed experimental evolution in Drosophila subobscura using selective regimes with slow or fast ramping protocols. Whereas both regimes showed increased heat tolerance with similar evolutionary rates, the correlated responses in thermal performance curves for locomotor behavior differed between selection regimes. These findings suggest that thermal rate and intensity may shape the evolution of correlated responses in other traits, urging the need to understand possible correlated responses at relevant levels such as life history and metabolism. In the present contribution, Mesas and Castañeda (2022) investigate whether the disparity in thermal performance curves observed in the previous experiment (Mesas et al 2021) could be explained by differences in metabolic energy production and consumption, and how this correlated with the reproductive output (fecundity and viability). Overall, the authors show some evidence for lowered enzyme activity and increased performance in life-history traits, particularly for the slow-ramping selected flies. Specifically, the authors observe a reduction in glucose metabolism and increased viability when evolving under slow ramping stress. Interestingly, both regimes show a general increase in fecundity, suggesting that adaptation to these higher temperatures is not costly (for reproduction) in the ancestral environment. The evidence for a somewhat lower metabolism in the slow-ramping lines suggests the evolution of a slow “pace of life”. The “pace of life” concept tries to bridge variation across several levels namely metabolism, physiology, behavior and life history, with low “pace of life” organisms presenting lower metabolic rates, later reproduction and higher longevity than fast “pace of life” organisms (Dammhahn et al 2018, Tuzun & Stocks 2022). As the authors state there is not a clear-cut association with the expectations of the pace of life hypothesis since there was evidence for increased reproductive output under both selection intensity regimes. This suggests that, given sufficient trait genetic variance, positively correlated responses may emerge during some stages of thermal evolution. As fecundity estimates in this study were focussed on early life, the possibility of a decrease in the cumulative reproductive output of the selected flies, even under benign conditions, cannot be excluded. This would help explain the apparent paradox of increased fecundity in selected lines. In this context, it would also be interesting to explore the variation in reproductive output at different temperatures, i.e to obtain thermal performance curves for life histories. Mesas and Castañeda (2022) raise important questions to pursue in the future and contribute to the growing evidence that, in order to predict the distribution of ectothermic species under current global warming conditions, we need to expand beyond determining the physiological thermal limits of each organism (Parratt et al 2021). Ultimately, integrating metabolic, life-history and behavioral changes during evolution under different thermal stresses within a coherent framework is key to developing better predictions of temperature effects on natural populations. Bennett, J.M., Sunday, J., Calosi, P. et al. The evolution of critical thermal limits of life on Earth. Nat Commun 12, 1198 (2021). https://doi.org/10.1038/s41467-021-21263-8 Dammhahn, M., Dingemanse, N.J., Niemelä, P.T. et al. Pace-of-life syndromes: a framework for the adaptive integration of behaviour, physiology and life history. Behav Ecol Sociobiol 72, 62 (2018). https://doi.org/10.1007/s00265-018-2473-y Mesas, A, Jaramillo, A, Castañeda, LE. Experimental evolution on heat tolerance and thermal performance curves under contrasting thermal selection in Drosophila subobscura. J Evol Biol 34, 767– 778 (2021). https://doi.org/10.1111/jeb.13777 Parratt, S.R., Walsh, B.S., Metelmann, S. et al. Temperatures that sterilize males better match global species distributions than lethal temperatures. Nat. Clim. Chang. 11, 481–484 (2021). https://doi.org/10.1038/s41558-021-01047-0 Santos, M, Castañeda, LE, Rezende, EL Keeping pace with climate change: what is wrong with the evolutionary potential of upper thermal limits? Ecology and evolution, 2(11), 2866-2880 (2012). https://doi.org/10.1002/ece3.385 Tüzün, N, Stoks, R. A fast pace-of-life is traded off against a high thermal performance. Proceedings of the Royal Society B, 289(1972), 20212414 (2022). https://doi.org/10.1098/rspb.2021.2414 Rezende, EL, Bozinovic, F, Szilágyi, A, Santos, M. Predicting temperature mortality and selection in natural Drosophila populations. Science, 369(6508), 1242-1245 (2020). https://doi.org/10.1126/science.aba9287 | Evolutionary responses of energy metabolism, development, and reproduction to artificial selection for increasing heat tolerance in Drosophila subobscura | Andres Mesas, Luis E. Castaneda | <p>Adaptations to warming conditions exhibited by ectotherms include increasing heat tolerance but also metabolic changes to reduce maintenance costs (metabolic depression), which can allow them to redistribute the energy surplus to biological fun... | | Adaptation, Evolutionary Ecology, Experimental Evolution, Life History | Inês Fragata | 2022-02-08 01:05:50 | ||
16 Dec 2016

POSTPRINT
Evolutionary robotics simulations help explain why reciprocity is rare in nature.Simulated robots and the evolution of reciprocityRecommended by Michael D Greenfield and Joël Meunier
Of the various forms of cooperative and altruistic behavior, reciprocity remains the most contentious. Humans certainly exhibit reciprocity – under certain circumstances – and various non-human animals behave in ways suggesting that they do as well. Thus, evolutionary biologists have sought to explain why non-relatives might engage in altruistic transactions when a substantial delay occurs between helping and compensation; i.e. an individual may be a donor today and a beneficiary tomorrow, or vice-versa. This quest, aided by game theory and computer modeling late in the past century, identified some strategies for reciprocal behavior that could work – in theory. But when biologists looked for confirmation of these strategies in animals they found little evidence that stood up to rigorous testing. In a recent paper André and Nolfi [1] offer a compelling reason for this observed rarity of reciprocity: Reciprocal behavior that animals might exhibit is a bit more complex than any of the game theoretic strategies, and even the simplest forms of realistic behavior would entail several nearly simultaneous mutations, an unlikely occurrence. André and Nolfi [1] relied on neural networks to test actors, robots that could evolve helping and reciprocal behavior from a basal level of selfishness. In an extensive series of simulations, they found that reciprocal behavior did not take hold in a population, largely because the various intermediates to full reciprocity were eliminated before the subsequent mutations occurred. The findings are satisfying given our current knowledge of animal behavior, but questions remain. Notably, how does one account for those rare cases in which reciprocity does meet all the criteria? The authors suggest some possibilities, but an analysis will await their next study. Reference [1] André J-B, Nolfi S. 2016. Evolutionary robotics simulations help explain why reciprocity is rare in nature. Scientific Reports 6:32785. doi: 10.1038/srep32785 | Evolutionary robotics simulations help explain why reciprocity is rare in nature. | André J-B, Nolfi S | The relative rarity of reciprocity in nature, contrary to theoretical predictions that it should be widespread, is currently one of the major puzzles in social evolution theory. Here we use evolutionary robotics to solve this puzzle. We show tha... | | Behavior & Social Evolution, Evolutionary Theory | Michael D Greenfield | 2016-12-16 18:08:31 | ||
22 Sep 2020
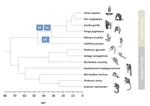
Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primatesStudying genetic antagonisms as drivers of genome evolutionRecommended by Mathieu Joron based on reviews by Qi Zhou and 3 anonymous reviewersSex chromosomes are special in the genome because they are often highly differentiated over much of their lengths and marked by degenerative evolution of their gene content. Understanding why sex chromosomes differentiate requires deciphering the forces driving their recombination patterns. Suppression of recombination may be subject to selection, notably because of functional effects of locking together variation at different traits, as well as longer-term consequences of the inefficient purge of deleterious mutations, both of which may contribute to patterns of differentiation [1]. As an example, male and female functions may reveal intrinsic antagonisms over the optimal genotypes at certain genes or certain combinations of interacting genes. As a result, selection may favour the recruitment of rearrangements blocking recombination and maintaining the association of sex-antagonistic allele combinations with the sex-determining locus. References [1] Charlesworth D (2017) Evolution of recombination rates between sex chromosomes. Philosophical Transactions of the Royal Society B: Biological Sciences, 372, 20160456. https://doi.org/10.1098/rstb.2016.0456 | Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primates | Rylan Shearn, Alison E. Wright, Sylvain Mousset, Corinne Régis, Simon Penel, Jean-François Lemaitre, Guillaume Douay, Brigitte Crouau-Roy, Emilie Lecompte, Gabriel A.B. Marais | <p>Sex chromosomes are typically comprised of a non-recombining region and a recombining pseudoautosomal region. Accurately quantifying the relative size of these regions is critical for sex chromosome biology both from a functional (i.e. number o... | | Bioinformatics & Computational Biology, Genome Evolution, Molecular Evolution, Reproduction and Sex, Sexual Selection | Mathieu Joron | 2019-02-04 15:16:32 | ||
03 Apr 2020

Evolution at two time-frames: ancient and common origin of two structural variants involved in local adaptation of the European plaice (Pleuronectes platessa)Genomic structural variants involved in local adaptation of the European plaiceRecommended by Maren Wellenreuther based on reviews by 3 anonymous reviewersAwareness has been growing that structural variants in the genome of species play a fundamental role in adaptive evolution and diversification [1]. Here, Le Moan and co-authors [2] report empirical genomic-wide SNP data on the European plaice (Pleuronectes platessa) across a major environmental transmission zone, ranging from the North Sea to the Baltic Sea. Regions of high linkage disequilibrium suggest the presence of two structural variants that appear to have evolved 220 kya. These two putative structural variants show weak signatures of isolation by distance when contrasted against the rest of the genome, but the frequency of the different putative structural variants appears to co-vary in some parts of the studied range with the environment, indicating the involvement of both selective and neutral processes. This study adds to the mounting body of evidence that structural genomic variants harbour significant information that allows species to respond and adapt to the local environmental context. References [1] Wellenreuther, M., Mérot, C., Berdan, E., & Bernatchez, L. (2019). Going beyond SNPs: the role of structural genomic variants in adaptive evolution and species diversification. Molecular ecology, 28(6), 1203-1209. doi: 10.1111/mec.15066 | Evolution at two time-frames: ancient and common origin of two structural variants involved in local adaptation of the European plaice (Pleuronectes platessa) | Alan Le Moan, Dorte Bekkevold & Jakob Hemmer-Hansen | <p>Changing environmental conditions can lead to population diversification through differential selection on standing genetic variation. Structural variant (SV) polymorphisms provide examples of ancient alleles that in time become associated with... | | Adaptation, Hybridization / Introgression, Population Genetics / Genomics, Speciation | Maren Wellenreuther | 2019-07-13 12:44:01 | ||
23 Jun 2021

Evolution of flowering time in a selfing annual plant: Roles of adaptation and genetic driftSeparating adaptation from drift: A cautionary tale from a self-fertilizing plantRecommended by Christoph Haag based on reviews by Pierre Olivier Cheptou, Jon Agren and Stefan LaurentIn recent years many studies have documented shifts in phenology in response to climate change, be it in arrival times in migrating birds, budset in trees, adult emergence in butterflies, or flowering time in annual plants (Coen et al. 2018; Piao et al. 2019). While these changes are, in part, explained by phenotypic plasticity, more and more studies find that they involve also genetic changes, that is, they involve evolutionary change (e.g., Metz et al. 2020). Yet, evolutionary change may occur through genetic drift as well as selection. Therefore, in order to demonstrate adaptive evolutionary change in response to climate change, drift has to be excluded as an alternative explanation (Hansen et al. 2012). A new study by Gay et al. (2021) shows just how difficult this can be. The authors investigated a recent evolutionary shift in flowering time by in a population an annual plant that reproduces predominantly by self-fertilization. The population has recently been subjected to increased temperatures and reduced rainfalls both of which are believed to select for earlier flowering times. They used a “resurrection” approach (Orsini et al. 2013; Weider et al. 2018): Genotypes from the past (resurrected from seeds) were compared alongside more recent genotypes (from more recently collected seeds) under identical conditions in the greenhouse. Using an experimental design that replicated genotypes, eliminated maternal effects, and controlled for microenvironmental variation, they found said genetic change in flowering times: Genotypes obtained from recently collected seeds flowered significantly (about 2 days) earlier than those obtained 22 generations before. However, neutral markers (microsatellites) also showed strong changes in allele frequencies across the 22 generations, suggesting that effective population size, Ne, was low (i.e., genetic drift was strong), which is typical for highly self-fertilizing populations. In addition, several multilocus genotypes were present at high frequencies and persisted over the 22 generations, almost as in clonal populations (e.g., Schaffner et al. 2019). The challenge was thus to evaluate whether the observed evolutionary change was the result of an adaptive response to selection or may be explained by drift alone. Here, Gay et al. (2021) took a particularly careful and thorough approach. First, they carried out a selection gradient analysis, finding that earlier-flowering plants produced more seeds than later-flowering plants. This suggests that, under greenhouse conditions, there was indeed selection for earlier flowering times. Second, investigating other populations from the same region (all populations are located on the Mediterranean island of Corsica, France), they found that a concurrent shift to earlier flowering times occurred also in these populations. Under the hypothesis that the populations can be regarded as independent replicates of the evolutionary process, the observation of concurrent shifts rules out genetic drift (under drift, the direction of change is expected to be random). The study may well have stopped here, concluding that there is good evidence for an adaptive response to selection for earlier flowering times in these self-fertilizing plants, at least under the hypothesis that selection gradients estimated in the greenhouse are relevant to field conditions. However, the authors went one step further. They used the change in the frequencies of the multilocus genotypes across the 22 generations as an estimate of realized fitness in the field and compared them to the phenotypic assays from the greenhouse. The results showed a tendency for high-fitness genotypes (positive frequency changes) to flower earlier and to produce more seeds than low-fitness genotypes. However, a simulation model showed that the observed correlations could be explained by drift alone, as long as Ne is lower than ca. 150 individuals. The findings were thus consistent with an adaptive evolutionary change in response to selection, but drift could only be excluded as the sole explanation if the effective population size was large enough. The study did provide two estimates of Ne (19 and 136 individuals, based on individual microsatellite loci or multilocus genotypes, respectively), but both are problematic. First, frequency changes over time may be influenced by the presence of a seed bank or by immigration from a genetically dissimilar population, which may lead to an underestimation of Ne (Wang and Whitlock 2003). Indeed, the low effective size inferred from the allele frequency changes at microsatellite loci appears to be inconsistent with levels of genetic diversity found in the population. Moreover, high self-fertilization reduces effective recombination and therefore leads to non-independence among loci. This lowers the precision of the Ne estimates (due to a higher sampling variance) and may also violate the assumption of neutrality due to the possibility of selection (e.g., due to inbreeding depression) at linked loci, which may be anywhere in the genome in case of high degrees of self-fertilization. There is thus no definite answer to the question of whether or not the observed changes in flowering time in this population were driven by selection. The study sets high standards for other, similar ones, in terms of thoroughness of the analyses and care in interpreting the findings. It also serves as a very instructive reminder to carefully check the assumptions when estimating neutral expectations, especially when working on species with complicated demographies or non-standard life cycles. Indeed the issues encountered here, in particular the difficulty of establishing neutral expectations in species with low effective recombination, may apply to many other species, including partially or fully asexual ones (Hartfield 2016). Furthermore, they may not be limited to estimating Ne but may also apply, for instance, to the establishment of neutral baselines for outlier analyses in genome scans (see e.g, Orsini et al. 2012). References Cohen JM, Lajeunesse MJ, Rohr JR (2018) A global synthesis of animal phenological responses to climate change. Nature Climate Change, 8, 224–228. https://doi.org/10.1038/s41558-018-0067-3 Gay L, Dhinaut J, Jullien M, Vitalis R, Navascués M, Ranwez V, Ronfort J (2021) Evolution of flowering time in a selfing annual plant: Roles of adaptation and genetic drift. bioRxiv, 2020.08.21.261230, ver. 4 recommended and peer-reviewed by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.08.21.261230 Hansen MM, Olivieri I, Waller DM, Nielsen EE (2012) Monitoring adaptive genetic responses to environmental change. Molecular Ecology, 21, 1311–1329. https://doi.org/10.1111/j.1365-294X.2011.05463.x Hartfield M (2016) Evolutionary genetic consequences of facultative sex and outcrossing. Journal of Evolutionary Biology, 29, 5–22. https://doi.org/10.1111/jeb.12770 Metz J, Lampei C, Bäumler L, Bocherens H, Dittberner H, Henneberg L, Meaux J de, Tielbörger K (2020) Rapid adaptive evolution to drought in a subset of plant traits in a large-scale climate change experiment. Ecology Letters, 23, 1643–1653. https://doi.org/10.1111/ele.13596 Orsini L, Schwenk K, De Meester L, Colbourne JK, Pfrender ME, Weider LJ (2013) The evolutionary time machine: using dormant propagules to forecast how populations can adapt to changing environments. Trends in Ecology & Evolution, 28, 274–282. https://doi.org/10.1016/j.tree.2013.01.009 Orsini L, Spanier KI, Meester LD (2012) Genomic signature of natural and anthropogenic stress in wild populations of the waterflea Daphnia magna: validation in space, time and experimental evolution. Molecular Ecology, 21, 2160–2175. https://doi.org/10.1111/j.1365-294X.2011.05429.x Piao S, Liu Q, Chen A, Janssens IA, Fu Y, Dai J, Liu L, Lian X, Shen M, Zhu X (2019) Plant phenology and global climate change: Current progresses and challenges. Global Change Biology, 25, 1922–1940. https://doi.org/10.1111/gcb.14619 Schaffner LR, Govaert L, De Meester L, Ellner SP, Fairchild E, Miner BE, Rudstam LG, Spaak P, Hairston NG (2019) Consumer-resource dynamics is an eco-evolutionary process in a natural plankton community. Nature Ecology & Evolution, 3, 1351–1358. https://doi.org/10.1038/s41559-019-0960-9 Wang J, Whitlock MC (2003) Estimating Effective Population Size and Migration Rates From Genetic Samples Over Space and Time. Genetics, 163, 429–446. PMID: 12586728 Weider LJ, Jeyasingh PD, Frisch D (2018) Evolutionary aspects of resurrection ecology: Progress, scope, and applications—An overview. Evolutionary Applications, 11, 3–10. https://doi.org/10.1111/eva.12563 | Evolution of flowering time in a selfing annual plant: Roles of adaptation and genetic drift | Laurène Gay, Julien Dhinaut, Margaux Jullien, Renaud Vitalis, Miguel Navascués, Vincent Ranwez, and Joëlle Ronfort | <p style="text-align: justify;">Resurrection studies are a useful tool to measure how phenotypic traits have changed in populations through time. If these traits modifications correlate with the environmental changes that occurred during the time ... | | Adaptation, Evolutionary Ecology, Genotype-Phenotype, Phenotypic Plasticity, Population Genetics / Genomics, Quantitative Genetics, Reproduction and Sex | Christoph Haag | 2020-08-21 17:26:59 | ||
17 Dec 2016
POSTPRINT
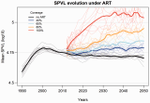
Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling studyPredicting HIV virulence evolution in response to widespread treatmentRecommended by Samuel Alizon and Roger Kouyos
It is a classical result in the virulence evolution literature that treatments decreasing parasite replication within the host should select for higher replication rates, thus driving increased levels of virulence if the two are correlated. There is some evidence for this in vitro but very little in the field. HIV infections in humans offer a unique opportunity to go beyond the simple predictions that treatments should favour more virulent strains because many details of this host-parasite system are known, especially the link between set-point virus load, transmission rate and virulence. To tackle this question, Herbeck et al. [1] used a detailed individual-based model. This is original because it allows them to integrate existing knowledge from the epidemiology and evolution of HIV (e.g. recent estimates of the ‘heritability’ of set-point virus load from one infection to the next). This detailed model allows them to formulate predictions regarding the effect of different treatment policies; especially regarding the current policy switch away from treatment initiation based on CD4 counts towards universal treatment. The results show that, perhaps as expected from the theory, treatments based on the level of remaining host target cells (CD4 T cells) do not affect virulence evolution because they do not strongly affect the virulence level that maximizes HIV’s transmission potential. However, early treatments can lead to moderate increase in virulence within several years if coverage is high enough. These results seem quite robust to variation of all the parameters in realistic ranges. The great step forward in this model is the ability to obtain quantitative prediction regarding how a virus may evolve in response to public health policies. Here the main conclusion is that given our current knowledge in HIV biology, the risk of virulence evolution is perhaps more limited than expected from a direct application of virulence evolution model. Interestingly, the authors also conclude that recently observed increased in HIV virulence [2-3] cannot be explained by the impact of antiretroviral therapy alone; which raises the question about the main mechanism behind this increase. Finally, the authors make the interesting suggestion that “changing virulence is amenable to being monitored alongside transmitted drug resistance in sentinel surveillance”. References [1] Herbeck JT, Mittler JE, Gottlieb GS, Goodreau SM, Murphy JT, Cori A, Pickles M, Fraser C. 2016. Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling study. Virus Evolution 2:vew028. doi: 10.1093/ve/vew028 [2] Herbeck JT, Müller V, Maust BS, Ledergerber B, Torti C, et al. 2012. Is the virulence of HIV changing? A meta-analysis of trends in prognostic markers of HIV disease progression and transmission. AIDS 26:193-205. doi: 10.1097/QAD.0b013e32834db418 [3] Pantazis N, Porter K, Costagliola D, De Luca A, Ghosn J, et al. 2014. Temporal trends in prognostic markers of HIV-1 virulence and transmissibility: an observational cohort study. Lancet HIV 1:e119-26. doi: 10.1016/s2352-3018(14)00002-2 | Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling study | Herbeck JT, Mittler JE, Gottlieb GS, Goodreau SM, Murphy JT, Cori A, Pickles M, Fraser C | There are global increases in the use of HIV antiretroviral therapy (ART), guided by clinical benefits of early ART initiation and the efficacy of treatment as prevention of transmission. Separately, it has been shown theoretically and empirically... | | Bioinformatics & Computational Biology, Evolutionary Applications, Evolutionary Epidemiology | Samuel Alizon | 2016-12-16 20:54:08 |




