
Latest recommendations

| Id | Title▲ | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
09 Nov 2018

Field evidence for manipulation of mosquito host selection by the human malaria parasite, Plasmodium falciparumMalaria host manipulation increases probability of mosquitoes feeding on humansRecommended by Alison Duncan based on reviews by Olivier Restif, Ricardo S. Ramiro and 1 anonymous reviewerParasites can manipulate their host’s behaviour to ensure their own transmission. These manipulated behaviours may be outside the range of ordinary host activities [1], or alter the crucial timing and/or location of a host’s regular activity. Vantaux et al show that the latter is true for the human malaria parasite, Plasmodium falciparum [2]. They demonstrate that three species of Anopheles mosquito were 24% more likely to choose human hosts, rather than other vertebrates, for their blood feed when they harboured transmissible stages (sporozoites) compared to when they were uninfected, or infected with non-transmissible malaria parasites [2]. Host choice is crucial for the malaria parasite Plasmodium falciparum to complete its life-cycle, as their host range is much narrower than the mosquito’s for feeding; P. falciparum can only develop in hominids, or closely related apes [3]. References [1] Thomas, F., Schmidt-Rhaesa, A., Martin, G., Manu, C., Durand, P., & Renaud, F. (2002). Do hairworms (Nematomorpha) manipulate the water seeking behaviour of their terrestrial hosts? Journal of Evolutionary Biology, 15(3), 356–361. doi: 10.1046/j.1420-9101.2002.00410.x | Field evidence for manipulation of mosquito host selection by the human malaria parasite, Plasmodium falciparum | Amelie Vantaux, Franck Yao, Domonbabele FdS Hien, Edwige Guissou, Bienvenue K Yameogo, Louis-Clement Gouagna, Didier Fontenille, Francois Renaud, Frederic Simard, Carlo Constantini, Frederic Thomas, Karine Mouline, Benjamin Roche, Anna Cohuet, Kou... | <p>Whether the malaria parasite *Plasmodium falciparum* can manipulate mosquito host choice in ways that enhance parasite transmission toward human is unknown. We assessed the influence of *P. falciparum* on the blood-feeding behaviour of three of... | | Evolutionary Ecology | Alison Duncan | 2018-02-28 09:12:14 | ||
16 Nov 2018

Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen IslandsIntrogression from related species reveals fine-scale structure in an isolated population of mussels and causes patterns of genetic-environment associationsRecommended by Marianne Elias based on reviews by Thomas Broquet and Tatiana GiraudAssessing population connectivity is central to understanding population dynamics, and is therefore of great importance in evolutionary biology and conservation biology. In the marine realm, the apparent absence of physical barriers, large population sizes and high dispersal capacities of most organisms often result in no detectable structure, thereby hindering inferences of population connectivity. In a review paper, Gagnaire et al. [1] propose several ideas to improve detection of population connectivity. Notably, using simulations they show that under certain circumstances introgression from one species into another may reveal cryptic population structure within that second species. References [1] Gagnaire, P.-A., Broquet, T., Aurelle, D., Viard, F., Souissi, A., Bonhomme, F., Arnaud-Haond, S., & Bierne, N. (2015). Using neutral, selected, and hitchhiker loci to assess connectivity of marine populations in the genomic era. Evolutionary Applications, 8, 769–786. doi: 10.1111/eva.12288 | Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen Islands | Christelle Fraïsse, Anne Haguenauer, Karin Gerard, Alexandra Anh-Thu Weber, Nicolas Bierne, Anne Chenuil | <p>Reticulated evolution -i.e. secondary introgression / admixture between sister taxa- is increasingly recognized as playing a key role in structuring infra-specific genetic variation and revealing cryptic genetic connectivity patterns. When admi... | | Hybridization / Introgression, Phylogeography & Biogeography, Population Genetics / Genomics | Marianne Elias | 2017-12-28 14:16:16 | ||
03 Aug 2017
POSTPRINT
Fisher's geometrical model and the mutational patterns of antibiotic resistance across dose gradientsWhat doesn’t kill us makes us stronger: can Fisher’s Geometric model predict antibiotic resistance evolution?Recommended by Inês Fragata and Claudia BankThe increasing number of reported cases of antibiotic resistance is one of today’s major public health concerns. Dealing with this threat involves understanding what drives the evolution of antibiotic resistance and investigating whether we can predict (and subsequently avoid or circumvent) it [1]. References [1] Palmer AC, and Kishony R. 2013. Understanding, predicting and manipulating the genotypic evolution of antibiotic resistance. Nature Review Genetics 14: 243—248. doi: 10.1038/nrg3351 [2] Tenaillon O. 2014. The utility of Fisher’s geometric model in evolutionary genetics. Annual Review of Ecology, Evolution and Systematics 45: 179—201. doi: 10.1146/annurev-ecolsys-120213-091846 [3] Blanquart F and Bataillon T. 2016. Epistasis and the structure of fitness landscapes: are experimental fitness landscapes compatible with Fisher’s geometric model? Genetics 203: 847—862. doi: 10.1534/genetics.115.182691 [4] Harmand N, Gallet R, Jabbour-Zahab R, Martin G and Lenormand T. 2017. Fisher’s geometrical model and the mutational patterns of antibiotic resistance across dose gradients. Evolution 71: 23—37. doi: 10.1111/evo.13111 [5] de Visser, JAGM, and Krug J. 2014. Empirical fitness landscapes and the predictability of evolution. Nature 15: 480—490. doi: 10.1038/nrg3744 [6] Palmer AC, Toprak E, Baym M, Kim S, Veres A, Bershtein S and Kishony R. 2015. Delayed commitment to evolutionary fate in antibiotic resistance fitness landscapes. Nature Communications 6: 1—8. doi: 10.1038/ncomms8385 | Fisher's geometrical model and the mutational patterns of antibiotic resistance across dose gradients | Noémie Harmand, Romain Gallet, Roula Jabbour-Zahab, Guillaume Martin, Thomas Lenormand | Fisher's geometrical model (FGM) has been widely used to depict the fitness effects of mutations. It is a general model with few underlying assumptions that gives a large and comprehensive view of adaptive processes. It is thus attractive in sever... | | Adaptation | Inês Fragata | 2017-08-01 16:06:02 | ||
18 Nov 2022
Fitness costs and benefits in response to artificial artesunate selection in PlasmodiumThe importance of understanding fitness costs associated with drug resistance throughout the life cycle of malaria parasitesRecommended by Silvie Huijben based on reviews by Sarah Reece and Marianna SzucsAntimalarial resistance is a major hurdle to malaria eradication efforts. The spread of drug resistance follows basic evolutionary principles, with competitive interactions between resistant and susceptible malaria strains being central to the fitness of resistant parasites. These competitive interactions can be used to design resistance management strategies, whereby a fitness cost of resistant parasites can be exploited through maintaining competitive suppression of the more fit drug-susceptible parasites. This can potentially be achieved using lower drug dosages or lower frequency of drug treatments. This approach has been demonstrated to work empirically in a rodent malaria model [1,2] and has been demonstrated to have clinical success in cancer treatments [3]. However, these resistance management approaches assume a fitness cost of the resistant pathogen, and, in the case of malaria parasites in general, and for artemisinin resistant parasites in particular, there is limited information on the presence of such fitness cost. The best suggestive evidence for the presence of fitness costs comes from the discontinuation of the use of the drug, which, in the case of chloroquine, was followed by a gradual drop in resistance frequency over the following decade [see e.g. 4,5]. However, with artemisinin derivative drugs still in use, alternative ways to study the presence of fitness costs need to be undertaken. References [1] Huijben S, Bell AS, Sim DG, Tomasello D, Mideo N, Day T, et al. 2013. Aggressive chemotherapy and the selection of drug resistant pathogens. PLoS Pathog. 9(9): e1003578. https://doi.org/10.1371/journal.ppat.1003578 [5] Mharakurwa S, Matsena-Zingoni Z, Mudare N, Matimba C, Gara TX, Makuwaza A, et al. 2021. Steep rebound of chloroquine-sensitive Plasmodium falciparum in Zimbabwe. J Infect Dis. 223(2): 306-9. https://doi.org/10.1093/infdis/jiaa368 | Fitness costs and benefits in response to artificial artesunate selection in Plasmodium | Villa M, Berthomieu A, Rivero A | <p style="text-align: justify;">Drug resistance is a major issue in the control of malaria. Mutations linked to drug resistance often target key metabolic pathways and are therefore expected to be associated with biological costs. The spread of dr... | | Evolutionary Applications, Life History | Silvie Huijben | 2022-01-31 13:01:16 | ||
28 Mar 2024
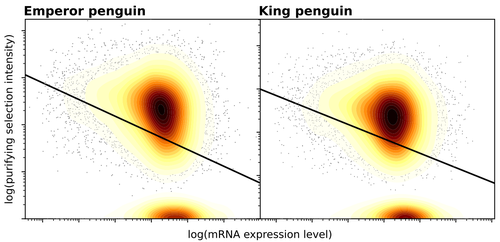
Gene expression is the main driver of purifying selection in large penguin populationsPurifying selection on highly expressed genes in PenguinsRecommended by Bruce Rannala based on reviews by Tanja Pyhäjärvi and 1 anonymous reviewerGiven the general importance of protein expression levels, in cells it is widely accepted that gene expression levels are often a target of natural selection and that most mutations affecting gene expression levels are therefore likely to be deleterious [1]. However, it is perhaps less obvious that the strength of selection on the regulated genes themselves may be influenced by their expression levels. This might be due to harmful effects of misfolded proteins, for example, when higher protein concentrations exist in cells [2]. Recent studies have suggested that highly expressed genes accumulate fewer deleterious mutations; thus a positive relationship appears to exist between gene expression levels and the relative strength of purifying selection [3]. The recommended paper by Trucchi et al. [4] examines the relationship between gene expression, purifying selection and a third variable -- effective population size -- in populations of two species of penguin with different population sizes, the Emperor penguin (Aptenodytes forsteri) and the King penguin (A. patagonicus). Using transcriptomic data and computer simulations modeling selection, they examine patterns of nonsynonymous and synonymous segregating polymorphisms (p) across genes in the two populations, concluding that even in relatively small populations purifying selection has an important effect in eliminating deleterious mutations. References 1] Gilad Y, Oshlack A, and Rifkin SA. 2006. Natural selection on gene expression. Trends in Genetics 22: 456-461. https://doi.org/10.1016/j.tig.2006.06.002 [4] Trucchi E, Massa P, Giannelli F, Latrille T, Fernandes FAN, Ancona L, Stenseth NC, Obiol JF, Paris J, Bertorelle G, and Le Bohec, C. 2023. Gene expression is the main driver of purifying selection in large penguin populations. bioRxiv 2023.08.08.552445, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.08.08.552445
| Gene expression is the main driver of purifying selection in large penguin populations | Emiliano Trucchi, Piergiorgio Massa, Francesco Giannelli, Thibault Latrille, Flavia A.N. Fernandes, Lorena Ancona, Nils Chr Stenseth, Joan Ferrer Obiol, Josephine Paris, Giorgio Bertorelle, Celine Le Bohec | <p style="text-align: justify;">Purifying selection is the most pervasive type of selection, as it constantly removes deleterious mutations arising in populations, directly scaling with population size. Highly expressed genes appear to accumulate ... | | Bioinformatics & Computational Biology, Evolutionary Dynamics, Evolutionary Theory, Population Genetics / Genomics | Bruce Rannala | 2023-08-09 17:53:03 | ||
31 Mar 2022
Gene network robustness as a multivariate characterGenetic and environmental robustness are distinct yet correlated evolvable traits in a gene networkRecommended by Frédéric Guillaume based on reviews by Diogo Melo, Charles Mullon and Charles Rocabert based on reviews by Diogo Melo, Charles Mullon and Charles Rocabert
Organisms often show robustness to genetic or environmental perturbations. Whether these two components of robustness can evolve separately is the focus of the paper by Le Rouzic [1]. Using theoretical analysis and individual-based computer simulations of a gene regulatory network model, he shows that multiple aspects of robustness can be investigated as a set of pleiotropically linked quantitative traits. While genetically correlated, various robustness components (e.g., mutational, developmental, homeostasis) of gene expression in the regulatory network evolved more or less independently from each other under directional selection. The quantitative approach of Le Rouzic could explain both how unselected robustness components can respond to selection on other components and why various robustness-related features seem to have their own evolutionary history. Moreover, he shows that all components were evolvable, but not all to the same extent. Robustness to environmental disturbances and gene expression stability showed the largest responses while increased robustness to genetic disturbances was slower. Interestingly, all components were positively correlated and remained so after selection for increased or decreased robustness. This study is an important contribution to the discussion of the evolution of robustness in biological systems. While it has long been recognized that organisms possess the ability to buffer genetic and environmental perturbations to maintain homeostasis (e.g., canalization [2]), the genetic basis and evolutionary routes to robustness and canalization are still not well understood. Models of regulatory gene networks have often been used to address aspects of robustness evolution (e.g., [3]). Le Rouzic [1] used a gene regulatory network model derived from Wagner’s model [4]. The model has as end product the expression level of a set of genes influenced by a set of regulatory elements (e.g., transcription factors). The level and stability of expression are a property of the regulatory interactions in the network. Le Rouzic made an important contribution to the study of such gene regulation models by using a quantitative genetics approach to the evolution of robustness. He crafted a way to assess the mutational variability and selection response of the components of robustness he was interested in. Le Rouzic’s approach opens avenues to investigate further aspects of gene network evolutionary properties, for instance to understand the evolution of phenotypic plasticity. Le Rouzic also discusses ways to measure his different robustness components in empirical studies. As the model is about gene expression levels at a set of protein-coding genes influenced by a set of regulatory elements, it naturally points to the possibility of using RNA sequencing to measure the variation of gene expression in know gene networks and assess their robustness. Robustness could then be studied as a multidimensional quantitative trait in an experimental setting. References [1] Le Rouzic, A (2022) Gene network robustness as a multivariate character. arXiv: 2101.01564, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://arxiv.org/abs/2101.01564 [2] Waddington CH (1942) Canalization of Development and the Inheritance of Acquired Characters. Nature, 150, 563–565. https://doi.org/10.1038/150563a0 [3] Draghi J, Whitlock M (2015) Robustness to noise in gene expression evolves despite epistatic constraints in a model of gene networks. Evolution, 69, 2345–2358. https://doi.org/10.1111/evo.12732 [4] Wagner A (1994) Evolution of gene networks by gene duplications: a mathematical model and its implications on genome organization. Proceedings of the National Academy of Sciences, 91, 4387–4391. https://doi.org/10.1073/pnas.91.10.4387 | Gene network robustness as a multivariate character | Arnaud Le Rouzic | <p style="text-align: justify;">Robustness to genetic or environmental disturbances is often considered as a key property of living systems. Yet, in spite of being discussed since the 1950s, how robustness emerges from the complexity of genetic ar... | | Bioinformatics & Computational Biology, Evolutionary Theory, Genotype-Phenotype, Quantitative Genetics | Frédéric Guillaume | Charles Mullon, Charles Rocabert, Diogo Melo | 2021-01-11 17:48:20 | |
12 Apr 2017

POSTPRINT
Genetic drift, purifying selection and vector genotype shape dengue virus intra-host genetic diversity in mosquitoesVectors as motors (of virus evolution)Recommended by Frédéric Fabre and Benoit MouryMany viruses are transmitted by biological vectors, i.e. organisms that transfer the virus from one host to another. Dengue virus (DENV) is one of them. Dengue is a mosquito-borne viral disease that has rapidly spread around the world since the 1940s. One recent estimate indicates 390 million dengue infections per year [1]. As many arthropod-borne vertebrate viruses, DENV has to cross several anatomical barriers in the vector, to multiply in its body and to invade its salivary glands before getting transmissible. As a consequence, vectors are not passive carriers but genuine hosts of the viruses that potentially have important effects on the composition of virus populations and, ultimately, on virus epidemiology and virulence. Within infected vectors, virus populations are expected to acquire new mutations and to undergo genetic drift and selection effects. However, the intensity of these evolutionary forces and the way they shape virus genetic diversity are poorly known. In their study, Lequime et al. [2] finely disentangled the effects of genetic drift and selection on DENV populations during their infectious cycle within mosquito (Aedes aegypti) vectors. They evidenced that the genetic diversity of viruses within their vectors is shaped by genetic drift, selection and vector genotype. The experimental design consisted in artificial acquisition of purified virus by mosquitoes during a blood meal. The authors monitored the diversity of DENV populations in Ae. aegypti individuals at different time points by high-throughput sequencing (HTS). They estimated the intensity of genetic drift and selection effects exerted on virus populations by comparing the DENV diversity at these sampling time points with the diversity in the purified virus stock (inoculum). Disentangling the effects of genetic drift and selection remains a methodological challenge because both evolutionary forces operate concomitantly and both reduce genetic diversity. However, selection reduces diversity in a reproducible manner among experimental replicates (here, mosquito individuals): the fittest variants are favoured at the expense of the weakest ones. In contrast, genetic drift reduces diversity in a stochastic manner among replicates. Genetic drift acts equally on all variants irrespectively of their fitness. The strength of genetic drift is frequently evaluated with the effective population size Ne: the lower Ne, the stronger the genetic drift [3]. The estimation of the effective population size of DENV populations by Lequime et al. [2] was based on single-nucleotide polymorphisms (SNPs) that were (i) present both in the inoculum and in the virus populations sampled at the different time points and (ii) that were neutral (or nearly-neutral) and therefore subjected to genetic drift only and insensitive to selection. As expected for viruses that possess small and constrained genomes, such neutral SNPs are extremely rare. Starting from a set of >1800 SNPs across the DENV genome, only three SNPs complied with the neutrality criteria and were enough represented in the sequence dataset for a precise Ne estimation. Using the method described by Monsion et al. [4], Lequime et al. [2] estimated Ne values ranging from 5 to 42 viral genomes (95% confidence intervals ranged from 2 to 161 founding viral genomes). Consequently, narrow bottlenecks occurred at the virus acquisition step, since the blood meal had allowed the ingestion of ca. 3000 infectious virus particles, on average. Interestingly, bottleneck sizes did not differ between mosquito genotypes. Monsion et al.’s [4] formula provides only an approximation of Ne. A corrected formula has been recently published [5]. We applied this exact Ne formula to the means and variances of the frequencies of the three neutral markers estimated before and after the bottlenecks (Table 1 in [2]), and nearly identical Ne estimates were obtained with both formulas. Selection intensity was estimated from the dN/dS ratio between the nonsynonymous and synonymous substitution rates using the HTS data on DENV populations. DENV genetic diversity increased following initial infection but was restricted by strong purifying selection during virus expansion in the midgut. Again, no differences were detected between mosquito genotypes. However and importantly, significant differences in DENV genetic diversity were detected among mosquito genotypes. As they could not be related to differences in initial genetic drift or to selection intensity, the authors raise interesting alternative hypotheses, including varying rates of de novo mutations due to differences in replicase fidelity or differences in the balancing selection regime. Interestingly, they also suggest that this observation could simply result from a methodological issue linked to the detection threshold of low-frequency SNPs. References [1] Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, et al. 2013. The global distribution and burden of dengue. Nature 496: 504–7 doi: 10.1038/nature12060 [2] Lequime S, Fontaine A, Gouilh MA, Moltini-Conclois I and Lambrechts L. 2016. Genetic drift, purifying selection and vector genotype shape dengue virus intra-host genetic diversity in mosquitoes. PloS Genetics 12: e1006111 doi: 10.1371/journal.pgen.1006111 [3] Charlesworth B. 2009. Effective population size and patterns of molecular evolution and variation. Nature Reviews Genetics 10: 195-205 doi: 10.1038/nrg2526 [4] Monsion B, Froissart R, Michalakis Y and Blanc S. 2008. Large bottleneck size in cauliflower mosaic virus populations during host plant colonization. PloS Pathogens 4: e1000174 doi: 10.1371/journal.ppat.1000174 [5] Thébaud G and Michalakis Y. 2016. Comment on ‘Large bottleneck size in cauliflower mosaic virus populations during host plant colonization’ by Monsion et al. (2008). PloS Pathogens 12: e1005512 doi: 10.1371/journal.ppat.1005512 | Genetic drift, purifying selection and vector genotype shape dengue virus intra-host genetic diversity in mosquitoes | Lequime S, Fontaine A, Gouilh MA, Moltini-Conclois I and Lambrechts L | Due to their error-prone replication, RNA viruses typically exist as a diverse population of closely related genomes, which is considered critical for their fitness and adaptive potential. Intra-host demographic fluctuations that stochastically re... | | Evolutionary Dynamics, Molecular Evolution, Population Genetics / Genomics | Frédéric Fabre | 2017-04-10 14:26:04 | ||
14 Dec 2023

Genetic sex determination in three closely related hydrothermal vent gastropods, including one species with intersex individualsA shared XY sex chromosome system with variable recombination ratesRecommended by Tanja Schwander based on reviews by Hugo Darras, Daniel Jeffries and 1 anonymous reviewerMany species with separate sexes have evolved sex chromosomes, with the sex-limited chromosomes (i.e. the Y or W chromosomes) exhibiting a wide range of genetic divergences from their homologous X or Z chromosomes (Bachtrog et al., 2014). Variable divergences can result from the cessation of recombination between sex chromosomes that occurred at different time points, with the mechanisms of initiation and expansion of recombination suppression along sex chromosomes remaining poorly understood (Charlesworth, 2017). The study by Castel et al (2023) describes the serendipitous discovery of a shared XY sex chromosome system in three closely related hydrothermal vent gastropods. The X and Y chromosomes appear to still recombine but at variable rates across the three species. This variation makes the gastropod system a very promising focus for future research on sex chromosome evolution. An additional intriguing finding is that some females in one of three gastropod species contain male reproductive tissue in their gonads, providing a fascinating case of a mixed or transitory sexual system. Overall, the study by Castel et al (2023) offers the first insights into the reproduction and sex chromosome system of animals living in deep marine vents, which have remained poorly studied and open outstanding research perspectives on these creatures. References Bachtrog, D., J.E.Mank, C.L.Peichel, M.Kirkpatrick, S.P.Otto, T.L. Ashman, M.W.Hahn, J.Kitano, I.Mayrose, R.Ming, et al. 2014.Sex determination: why so many ways of doing it? PLoSBiol. 12:e1001899. https://doi.org/10.1371/journal.pbio.1001899 Charlesworth, D. Young sex chromosomes in plants and animals. 2019. New Phytologist 224: 1095–1107. https://doi.org/10.1111/nph.16002 Castel J, Pradillon F, Cueff V, Leger G, Daguin-Thiébaut C, Ruault S, Mary J, Hourdez S, Jollivet D, and Broquet T 2023. Genetic sex determination in three closely related hydrothermal vent gastropods, including one species with intersex individuals. bioRxiv, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.04.11.536409 | Genetic sex determination in three closely related hydrothermal vent gastropods, including one species with intersex individuals | Castel J, Pradillon F, Cueff V, Leger G, Daguin-Thiébaut C, Ruault S, Mary J, Hourdez S, Jollivet D, and Broquet T | <p style="text-align: justify;">Molluscs have a wide variety of sexual systems and have undergone many transitions from separate sexes to hermaphroditism or vice versa, which is of interest for studying the evolution of sex determination and diffe... | | Population Genetics / Genomics, Reproduction and Sex | Tanja Schwander | 2023-04-14 11:48:25 | ||
08 Feb 2019
Genome plasticity in Papillomaviruses and de novo emergence of E5 oncogenesE5, the third oncogene of PapillomavirusRecommended by Hirohisa Kishino based on reviews by Leonardo de Oliveira Martins and 1 anonymous reviewerPapillomaviruses (PVs) infect almost all mammals and possibly amniotes and bony fishes. While most of them have no significant effects on the hosts, some induce physical lesions. Phylogeny of PVs consists of a few crown groups [1], among which AlphaPVs that infect primates including human have been well studied. They are associated to largely different clinical manifestations: non-oncogenic PVs causing anogenital warts, oncogenic and non-oncogenic PVs causing mucosal lesions, and non-oncogenic PVs causing cutaneous warts. References [1] Bravo, I. G., & Alonso, Á. (2004). Mucosal human papillomaviruses encode four different E5 proteins whose chemistry and phylogeny correlate with malignant or benign growth. Journal of virology, 78, 13613-13626. doi: 10.1128/JVI.78.24.13613-13626.2004 | Genome plasticity in Papillomaviruses and de novo emergence of E5 oncogenes | Anouk Willemsen, Marta Félez-Sánchez, and Ignacio G. Bravo | <p>The clinical presentations of papillomavirus (PV) infections come in many different flavors. While most PVs are part of a healthy skin microbiota and are not associated to physical lesions, other PVs cause benign lesions, and only a handful of ... | | Genome Evolution, Molecular Evolution, Phylogenetics / Phylogenomics | Hirohisa Kishino | 2018-06-04 16:15:39 | ||
10 Jan 2019

Genomic data provides new insights on the demographic history and the extent of recent material transfers in Norway spruceDisentangling the recent and ancient demographic history of European spruce speciesRecommended by Jason Holliday based on reviews by 1 anonymous reviewerGenetic diversity in temperate and boreal forests tree species has been strongly affected by late Pleistocene climate oscillations [2,3,5], but also by anthropogenic forces. Particularly in Europe, where a long history of human intervention has re-distributed species and populations, it can be difficult to know if a given forest arose through natural regeneration and gene flow or through some combination of natural and human-mediated processes. This uncertainty can confound inferences of the causes and consequences of standing genetic variation, which may impact our interpretation of demographic events that shaped species before humans became dominant on the landscape. In their paper entitled "Genomic data provides new insights on the demographic history and the extent of recent material transfers in Norway spruce", Chen et al. [1] used a genome-wide dataset of 400k SNPs to infer the demographic history of Picea abies (Norway spruce), the most widespread and abundant spruce species in Europe, and to understand its evolutionary relationship with two other spruces (Picea obovata [Siberian spruce] and P. omorika [Serbian spruce]). Three major Norway spruce clusters were identified, corresponding to central Europe, Russia and the Baltics, and Scandinavia, which agrees with previous studies. The density of the SNP data in the present paper enabled inference of previously uncharacterized admixture between these groups, which corresponds to the timing of postglacial recolonization following the last glacial maximum (LGM). This suggests that multiple migration routes gave rise to the extant distribution of the species, and may explain why Chen et al.'s estimates of divergence times among these major Norway spruce groups were older (15mya) than those of previous studies (5-6mya) – those previous studies may have unknowingly included admixed material [4]. Treemix analysis also revealed extensive admixture between Norway and Siberian spruce over the last ~100k years, while the geographically-restricted Serbian spruce was both isolated from introgression and had a dramatically smaller effective population size (Ne) than either of the other two species. This small Ne resulted from a bottleneck associated with the onset of the iron age ~3000 years ago, which suggests that anthropogenic depletion of forest resources has severely impacted this species. Finally, ancestry of Norway spruce samples collected in Sweden and Denmark suggest their recent transfer from more southern areas of the species range. This northward movement of genotypes likely occurred because the trees performed well relative to local provenances, which is a common observation when trees from the south are planted in more northern locations (although at the potential cost of frost damage due to inappropriate phenology). While not the reason for the transfer, the incorporation of southern seed sources into the Swedish breeding and reforestation program may lead to more resilient forests under climate change. Taken together, the data and analysis presented in this paper allowed inference of the intra- and interspecific demographic histories of a tree species group at a very high resolution, and suggest caveats regarding sampling and interpretation of data from areas with a long history of occupancy by humans. References [1] Chen, J., Milesi, P., Jansson, G., Berlin, M., Karlsson, B., Aleksić, J. M., Vendramin, G. G., Lascoux, M. (2018). Genomic data provides new insights on the demographic history and the extent of recent material transfers in Norway spruce. BioRxiv, 402016. ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/402016 | Genomic data provides new insights on the demographic history and the extent of recent material transfers in Norway spruce | Jun Chen, Lili Li, Pascal Milesi, Gunnar Jansson, Mats Berlin, Bo Karlsson, Jelena Aleksic, Giovanni G Vendramin, Martin Lascoux | <p>Primeval forests are today exceedingly rare in Europe and transfer of forest reproductive material for afforestation and improvement have been very common, especially over the last two centuries. This can be a serious impediment when inferring ... | | Evolutionary Applications, Hybridization / Introgression, Population Genetics / Genomics | Jason Holliday | Anonymous, Anonymous | 2018-08-29 08:33:15 |




