
Latest recommendations

| Id | Title | Authors▼ | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
30 Jun 2023
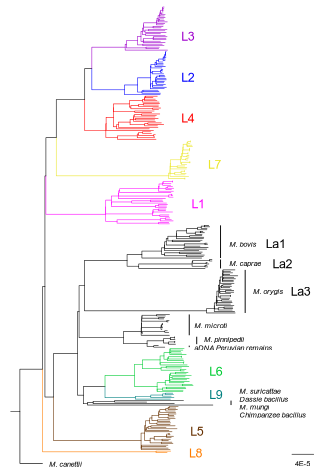
How do monomorphic bacteria evolve? The Mycobacterium tuberculosis complex and the awkward population genetics of extreme clonalityHow the tubercle bacillus got its genome: modernising, modelling, and making sense of the stories we tellRecommended by B. Jesse Shapiro based on reviews by 2 anonymous reviewersIn this instructive review, Stritt and Gagneux offer a balanced perspective on the evolutionary forces shaping Mycobacterium tuberculosis and make the case that our instinct for storytelling be balanced with quantitative models. M. tuberculosis is perhaps the best-known clonal bacterial pathogen – evolving largely in the absence of horizontal gene transfer. Its genome is full of puzzling patterns, including much higher GC content than most intracellular pathogens (which suggests efficient selection to resist AT-skewed mutational bias) but a very high ratio of nonsynonymous to synonymous substitution rates (dN/dS ~ 0.5, typically interpreted as weak selection against deleterious amino acid changes). The authors offer alternative explanations for these patterns, framing the question: is M. tuberculosis evolution shaped mainly by drift or by efficient selection? They propose that this question can only be answered by accounting for the pathogen’s extreme clonality. A clonal lifestyle can have its advantages, for example when adaptations must arise in a particular order (Kondrashov and Kondrashov 2001). An important disadvantage highlighted by the authors are linkage effects: without recombination to shuffle them up, beneficial mutations are linked to deleterious mutations in the same genome (hitchhiking) and purging deleterious mutations also purges neutral diversity across the genome (background selection). The authors propose the latter – efficient purifying selection and strong linkage – as an explanation for the low genetic diversity observed in M. tuberculosis. This is of course not exclusive of other related explanations, such as clonal interference (Gerrish and Lenski 1998). They also champion the use of forward evolutionary simulations (Haller and Messer 2019) to model the interplay between selection, recombination, and demography as a powerful alternative to traditional backward coalescent models. At times, Stritt and Gagneux are pessimistic about our existing methods – arguing that dN/dS and homoplasies “tell us little about the frequency and strength of selection.” Even though I favour a more optimistic view, I fully agree that our traditional population genetic metrics are sensitive to a slew of different deviations from a standard neutral evolution model and must be interpreted with caution. As I and others have argued, the extent of recombination (measured as the amount of linkage in a genome) is a key factor in determining how best to test for natural selection (Shapiro et al. 2009) and to conduct genotype-phenotype association studies (Chen and Shapiro 2021) in microbes. While this article is focused on the well-studied M. tuberculosis complex, there are many parallels with other clonal bacteria, including pathogens and symbionts. Whatever your favourite bug, we must all be careful to make sure the stories we tell about them are not “just so tales” but are supported, to the extent possible, by data and quantitative models. References Chen, Peter E., and B. Jesse Shapiro. 2021. "Classic Genome-Wide Association Methods Are Unlikely to Identify Causal Variants in Strongly Clonal Microbial Populations." bioRxiv. Stritt, C., Gagneux, S. (2023). How do monomorphic bacteria evolve? The Mycobacterium tuberculosis complex and the awkward population genetics of extreme clonality. EcoEvoRxiv, ver.3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.32942/X2GW2P | How do monomorphic bacteria evolve? The *Mycobacterium tuberculosis* complex and the awkward population genetics of extreme clonality | Christoph Stritt, Sebastien Gagneux | <p style="text-align: justify;">Exchange of genetic material through sexual reproduction or horizontal gene transfer is ubiquitous in nature. Among the few outliers that rarely recombine and mainly evolve by <em>de novo</em> mutation are a group o... | | Evolutionary Dynamics, Genome Evolution, Molecular Evolution, Population Genetics / Genomics, Reproduction and Sex | B. Jesse Shapiro | Gonçalo Themudo | 2022-12-16 13:41:53 | |
16 Nov 2018

Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen IslandsIntrogression from related species reveals fine-scale structure in an isolated population of mussels and causes patterns of genetic-environment associationsRecommended by Marianne Elias based on reviews by Thomas Broquet and Tatiana GiraudAssessing population connectivity is central to understanding population dynamics, and is therefore of great importance in evolutionary biology and conservation biology. In the marine realm, the apparent absence of physical barriers, large population sizes and high dispersal capacities of most organisms often result in no detectable structure, thereby hindering inferences of population connectivity. In a review paper, Gagnaire et al. [1] propose several ideas to improve detection of population connectivity. Notably, using simulations they show that under certain circumstances introgression from one species into another may reveal cryptic population structure within that second species. References [1] Gagnaire, P.-A., Broquet, T., Aurelle, D., Viard, F., Souissi, A., Bonhomme, F., Arnaud-Haond, S., & Bierne, N. (2015). Using neutral, selected, and hitchhiker loci to assess connectivity of marine populations in the genomic era. Evolutionary Applications, 8, 769–786. doi: 10.1111/eva.12288 | Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen Islands | Christelle Fraïsse, Anne Haguenauer, Karin Gerard, Alexandra Anh-Thu Weber, Nicolas Bierne, Anne Chenuil | <p>Reticulated evolution -i.e. secondary introgression / admixture between sister taxa- is increasingly recognized as playing a key role in structuring infra-specific genetic variation and revealing cryptic genetic connectivity patterns. When admi... | | Hybridization / Introgression, Phylogeography & Biogeography, Population Genetics / Genomics | Marianne Elias | 2017-12-28 14:16:16 | ||
02 Sep 2022

Introgression between highly divergent sea squirt genomes: an adaptive breakthrough?A match made in the Anthropocene: human-mediated adaptive introgression across a speciation continuumRecommended by Fernando Racimo based on reviews by Michael Westbury, Andrew Foote and Erin CalfeeThe long-distance transport and introduction of new species by humans is increasingly leading divergent lineages to interact, and sometimes interbreed, even after thousands or millions of years of separation. It is thus of prime importance to understand the consequences of these contemporary admixture events on the evolutionary fitness of interacting organisms, and their ecological implications. Ciona robusta and Ciona intestinalis are two species of sea squirts that diverged between 1.5 and 2 million years ago and recently came into contact again. This occurred through human-mediated introduction of C. robusta (native to the Northwest Pacific) into the range of C. intestinalis (the English channeled Northeast Atlantic). In this study, Fraïsse et al. (2022) follow up on earlier work by Le Moan et al. (2021), who had identified a long genomic hotspot of introgression of C. robusta ancestry segments in chromosome 5 of C. intestinalis. The hotspot bears suggestive evidence of positive selection and the authors aimed to investigate this further using fully phased whole-genome sequences. The authors narrow down on the exact boundaries of the introgressed region, and make a compelling case that it has been the likely target of positive selection after introgression, using various complementary approaches based on patterns of population differentiation, haplotype structure and local levels of diversity in the region. Using extensive demographic modeling, they also show that the introgression event was likely recent (approximately 75 years ago), and distinct from other tracts in the C. intestinalis genome that are likely a product of more ancient episodes of interbreeding in the past 30,000 years. Narrowing down on the potential drivers of selection, the authors show that candidate SNPs in the region overlap with the cytochrome family 2 subfamily U gene - involved in the detoxification of exogenous compounds - potentially reflecting adaptation to chemicals encountered in the sea squirt's environment. There also appears to be copy number variation at the candidate SNPs, which provides clues into the adaptation mechanism in the region. All reviewers agreed that the work carried out by the authors is elegant and the results are robustly supported and well presented. In a round of reviews, various clarifications of the manuscript were suggested by the reviewers, including on the quality of the newly generated sequencing data, and some suggestions for qualifications on the conclusions reached by the authors as well as changes in the figures to increase their clarity. The authors addressed the different concerns of the reviewers, and the new version is much improved. This study into human-mediated introgression and its consequences for adaptation is, in my view, both well thought-out and executed. I therefore provide an enthusiastic recommendation of this manuscript. References Fraïsse C, Le Moan A, Roux C, Dubois G, Daguin-Thiébaut C, Gagnaire P-A, Viard F and Bierne N (2022) Introgression between highly divergent sea squirt genomes: an adaptive breakthrough? bioRxiv, 2022.03.22.485319, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.03.22.485319 Le Moan A, Roby C, Fraïsse C, Daguin-Thiébaut C, Bierne N, Viard F (2021) An introgression breakthrough left by an anthropogenic contact between two ascidians. Molecular Ecology, 30, 6718–6732. https://doi.org/10.1111/mec.16189 | Introgression between highly divergent sea squirt genomes: an adaptive breakthrough? | Christelle Fraïsse, Alan Le Moan, Camille Roux, Guillaume Dubois, Claire Daguin-Thiébaut, Pierre-Alexandre Gagnaire, Frédérique Viard, Nicolas Bierne | <p style="text-align: justify;">Human-mediated introductions are reshuffling species distribution on a global scale. Consequently, an increasing number of allopatric taxa are now brought into contact, promoting introgressive hybridization between ... | | Adaptation, Hybridization / Introgression, Population Genetics / Genomics | Fernando Racimo | 2022-04-14 15:30:42 | ||
11 Apr 2023
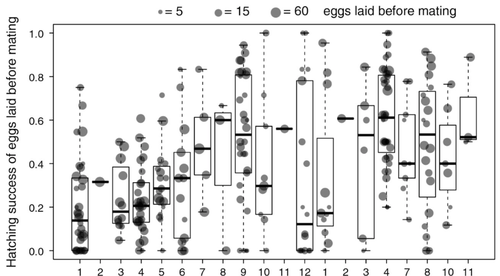
Facultative parthenogenesis: a transient state in transitions between sex and obligate asexuality in stick insects?Facultative parthenogenesis and transitions from sexual to asexual reproductionRecommended by Trine Bilde based on reviews by 3 anonymous reviewers based on reviews by 3 anonymous reviewers
Despite a vast array of ways in which organisms can reproduce (Bell, 1982), most animals engage in sexual reproduction (Otto & Lenormand, 2002). A fascinating alternative to sex is parthenogenesis, where offspring are produced asexually from a gamete, typically the egg, without receiving genetic material from another gamete (Simon, Delmotte, Rispe, & Crease, 2003). One of the long-standing questions in the field is why parthenogenesis is not more widespread, given the costs associated with sex (Otto & Lenormand, 2002). Natural populations of most species appear to be reproducing either sexually or parthenogenetically, even if a species can employ both reproductive modes (Larose et al 2023). Larose et al (2023) highlight the conundrum in this pattern, as organisms that are capable of employing parthenogenesis facultatively would be able to gain the benefits of both modes of reproduction. Why then, is facultative parthenogenesis not more common? Larose et al (2023) propose that constraints on being efficient in both sexual and asexual reproduction could cause a trade-off between reproductive modes that favours an obligate strategy of either sex or no sex. This would provide an explanation for why facultative parthenogenesis is rare. Timema stick insects provide an excellent system to investigate reproductive strategies, as some species have parthenogenetic females, while other species are sexual, and they show repeated transitions from sexual reproduction to obligate parthenogenesis (Schwander & Crespi, 2009). The authors performed comprehensive and complementary studies in a recently discovered species T. douglasi, in which populations show both modes of reproduction, with some populations consisting only of females and others showing equal proportions of males and females. The sex ratio varied significantly, with the proportion of females ranging between 43-100% across 29 populations. These populations form a monophyletic clade with clustering into three genetic lineages and only a few cases of admixture. Females from all populations were capable of producing unfertilized eggs, but the hatching success varied hugely among populations and lineages (3-100%). Parthenogenetically produced offspring were homozygous, showing that parthenogenesis causes a complete loss of heterozygosity in a single generation. After producing eggs as virgins, females were mated to assess the capacity to also reproduce sexually, and fertilization increased the hatching success of eggs in two lineages. In one lineage, in which the hatching success of unfertilized eggs is similar to that of other sexually reproducing Timema species, fertilization reduced egg-hatching success, indicating a trade-off between reproductive modes with parthenogenetic reproduction performing best. Approximately 58% of the offspring produced after mating were fertilized, demonstrating the capacity of females to reproduce parthenogenetically also after mating has occurred, however with huge variation among individuals. This wonderful and meticulously performed study produces strong and complementary evidence for facultative parthenogenesis in T. douglasi populations. The study shows large variation in how reproductive mode is employed, supporting the existence of a trade-off between sexual and parthenogenetic reproduction. This might be an example of an ongoing transition from sexual to asexual reproduction, which indicates that obligate parthenogenesis may derive via transient facultative parthenogenesis. REFERENCES Bell, G. (1982) The Masterpiece of Nature: The Evolution and Genetics of Sexuality. University of California Press. 635 p. Otto, S. P., & Lenormand, T. (2002). Resolving the paradox of sex and recombination. Nature Reviews Genetics, 3(4), 252-261. https://doi.org/10.1038/nrg761 Schwander, T., & Crespi, B. J. (2009). Multiple direct transitions from sexual reproduction to apomictic parthenogenesis in Timema stick insects. Evolution, 63(1), 84-103. Simon, J.-C., Delmotte, F., Rispe, C., & Crease, T. (2003). Phylogenetic relationships between parthenogens and their sexual relatives: the possible routes to parthenogenesis in animals. Biological Journal of the Linnean Society, 79(1), 151-163. https://doi.org/10.1046/j.1095-8312.2003.00175.x Larose, C., Lavanchy, G., Freitas, S., Parker, D.J., Schwander, T. (2023) Facultative parthenogenesis: a transient state in transitions between sex and obligate asexuality in stick insects? bioRxiv, 2022.03.25.485836, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.03.25.485836 | Facultative parthenogenesis: a transient state in transitions between sex and obligate asexuality in stick insects? | Chloé Larose, Guillaume Lavanchy, Susana Freitas, Darren J. Parker, Tanja Schwander | <p>Transitions from obligate sex to obligate parthenogenesis have occurred repeatedly across the tree of life. Whether these transitions occur abruptly or via a transient phase of facultative parthenogenesis is rarely known. We discovered and char... | | Reproduction and Sex | Trine Bilde | 2022-05-20 10:41:13 | ||
06 Mar 2023
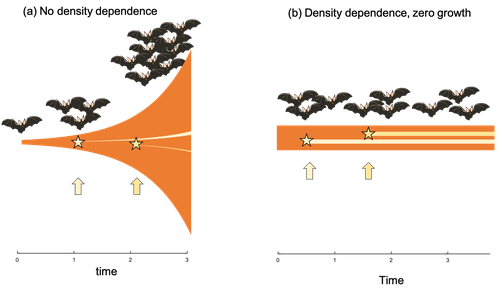
Extrinsic mortality and senescence: a guide for the perplexedGetting old gracefully, and risk of dying before getting there: a new guide to theory on extrinsic mortality and senescenceRecommended by Sinead English and Shinichi Nakagawa based on reviews by Nicole Walasek and 1 anonymous reviewerWhy is there such variation across species and populations in the rate at which individuals show wear and tear as they get older? Several compelling theoretical explanations have been developed on the conditions under which selection allows for or prevents senescence; a notable one being that proposed by George C Williams in 1957 based on the idea of antagonistic pleiotropy (Williams, 1957). One of the testable predictions of this theory is that, in populations where adults experience higher mortality, senescence is expected to be faster. This is one of the most influential predictions of the paper, being intuitive (when individuals are less likely to survive to later age classes, we expect weakened selection on traits that would avoid senescence in these classes), and fitting with ‘live fast, die young’ life history framing. As such, it has been widely incorporated into how we think about the evolution of senescence and has received considerable support in comparative studies across species and populations. However, it would be misleading to sit back at this point and think we have ‘solved’ the problem of understanding variation in senescence, and how this is linked with mortality. It turns out that the Williams 1957 paper is hotly contested by theoreticians: for the past 30 years – with increasing focus in the last 4 years – a growing body of models and opinion pieces have proposed flaws in the paper itself and in how it has been interpreted (Abrams, 1993; André and Rousset, 2020; Day and Abrams, 2020; Moorad et al., 2019). Central to several of these critiques is that explicit consideration of density dependence (not considered in Williams’ original paper) changes the conditions under which his predictions hold. A new preprint by de Vries, Gallipaud and Kokko brings further clarity to such critiques of the original paper (Vries et al., 2023). Beyond just continuing the tradition of critiquing Williams’ prediction, however, de Vries et al. provide a clear guide that is accessible to non-theoreticians about the issues with William’s prediction, and the way in which density dependence and how it operates can change when we expect senescence to occur. Rather than reiterate their points here, we suggest a close reading of the paper itself, along with an excellent overview of the paper in a recent blog by Daniel Nettle (Nettle, 2022). In brief, the paper starts by synthesizing earlier theoretical and empirical studies on the topic and presenting a very simple model to highlight how – in the absence of density dependence – Williams’ prediction does not hold because of the unregulated population growth, which is necessarily higher when there is low mortality. As a result, for a lineage with low mortality, any fitness advantage of placing offspring into the lineage later (i.e. selection for reduced senescence) is exactly cancelled out by the fact that earlier-produced offspring have higher fitness returns. They then present a more complex framework, which incorporates realistic mortality distributions, trade-offs between survival and reproduction, and use a series of 10 scenarios of density dependence (and whether this acts on survival or fecundity, and also whether it depends on a threshold or stochastic, or exerts continuing pressure on the trait) to explore selection on fitness-associated traits with age depending on extrinsic mortality. This then generates a range of results for when the Williams prediction holds, when there is no link between mortality and senescence, and when there is an ‘anti-Williams’ result – i.e., where senescence is slower when there is a high mortality. As has been shown in earlier studies, density dependence and how it operates matters, and Williams’ prediction holds most when density dependence affects juvenile age classes (in their model, when adults are less likely to produce them – i.e. there is density dependence on fecundity; or when there is less recruitment into the adult population due to, for example, competition among juveniles). So, perhaps we are now at a point where we can lay to rest the debate on the issues specifically with Williams’ original paper, and instead consider more broadly the key factors to measure when predicting patterns of senescence, and what is tangible for empiricists to quantify in their studies. Here, de Vries et al. provide some helpful insights both into the limitations of their approach and what modelling should be done moving forward, and into how we can link existing studies (for example comparing senescence among populations with varying predation pressure) to the theoretical predictions. At the heart of the latter is understanding the mechanism of density-dependent regulation – does it affect survival or fecundity, which age classes are most sensitive, and how do key traits depend on density? – and this is often difficult to measure in field studies. And from all this we can learn that even very intuitive and extensively discussed concepts in biology do not always have as firm theoretical underpinnings as assumed, and – as should not be surprising – biology is complex and rather than one clear pattern being predicted, this can depend on a multitude of factors. REFERENCES Abrams PA (1993) Does increased mortality favor the evolution of more rapid senescence? Evolution, 47, 877–887. https://doi.org/10.1111/j.1558-5646.1993.tb01241.x André J-B, Rousset F (2020) Does extrinsic mortality accelerate the pace of life? A bare-bones approach. Evolution and Human Behavior, 41, 486–492. https://doi.org/10.1016/j.evolhumbehav.2020.03.002 Day T, Abrams PA (2020) Density Dependence, Senescence, and Williams’ Hypothesis. Trends in Ecology & Evolution, 35, 300–302. https://doi.org/10.1016/j.tree.2019.11.005 Moorad J, Promislow D, Silvertown J (2019) Evolutionary Ecology of Senescence and a Reassessment of Williams’ ‘Extrinsic Mortality’ Hypothesis. Trends in Ecology & Evolution, 34, 519–530. https://doi.org/10.1016/j.tree.2019.02.006 Nettle AD (2022) Live fast and die young (maybe). https://www.danielnettle.org.uk/2022/02/18/live-fast-and-die-young-maybe/ (accessed 2.27.23). de Vries C, Galipaud M, Kokko H (2023) Extrinsic mortality and senescence: a guide for the perplexed. bioRxiv, 2022.01.27.478060, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.01.27.478060 Williams GC (1957) Pleiotropy, natural selection, and the evolution of senescence. Evolution, 11, 398–411. https://doi.org/10.1111/j.1558-5646.1957.tb02911.x | Extrinsic mortality and senescence: a guide for the perplexed | Charlotte de Vries, Matthias Galipaud, Hanna Kokko | <p style="text-align: justify;">Do environments or species traits that lower the mortality of individuals create selection for delaying senescence? Reading the literature creates an impression that mathematically oriented biologists cannot agree o... | | Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Theory, Life History | Sinead English | 2022-08-26 14:30:16 | ||
07 Nov 2017

MaxTiC: Fast ranking of a phylogenetic tree by Maximum Time Consistency with lateral gene transfersDating nodes in a phylogeny using inferred horizontal gene transfersRecommended by Tatiana Giraud and Toni Gabaldon based on reviews by Alexandros Stamatakis, Mukul Bansal and 2 anonymous reviewersDating nodes in a phylogeny is an important problem in evolution and is typically performed by using molecular clocks and fossil age estimates [1]. The manuscript by Chauve et al. [2] reports a novel method, which uses lateral gene transfers to help ordering nodes in a species tree. The idea is that a lateral gene transfer can only occur between two species living at the same time, which indirectly informs on node relative ages in a phylogeny: the donor species cannot be more recent than the recipient species. Horizontal gene transfers are increasingly recognized as frequent, even in eukaryotes, and especially in micro-organisms that have little fossil records [3-7]. Yet, such an important source of information has been very rarely used so far for inferring relative node ages in phylogenies. In this context, the method by Chauve et al. [2] represents an innovative and original approach to a difficult problem. An obvious limitation of the approach is that it relies on inferences of horizontal transfers, which detection is in itself a difficult problem. Incomplete taxon sampling, or the extinction of the true donor lineage may render patterns difficult to interpret in a temporary fashion. Yet, for clades with no fossils this may be the only piece of information we have at hand, and the growing amount of sequence data is likely to minimize issues derived from incomplete sampling. The developed method, MaxTiC (for Maximal Time Consistency) [2], represents a very nice application of theoretical developments on the well-known « Feedback Arc Set » computer science problem to the evolutionary question of ordering nodes in a phylogeny. MaxTiC uses as input a species tree and a set of time constraints based on lateral gene transfers inferred using other softwares, and minimizes conflicts between node ordering and these time constraints. The application of MaxTiC on simulated datasets indicated that node ordering was fairly accurate [2]. MaxTiC is implemented in a freely available software, which represents original and relevant contribution to the field of evolutionary biology. References [1] Donoghue P and Smith M, editors. 2003. Telling the evolutionary time. CRC press. [2] Chauve C, Rafiey A, Davin AA, Scornavacca C, Veber P, Boussau B, Szöllősi GJ, Daubin V and Tannier E. 2017. MaxTiC: Fast ranking of a phylogenetic tree by Maximum Time Consistency with lateral gene transfers. bioRxiv 127548, ver. 6 of 6th November 2017. doi: 10.1101/127548 [3] Ropars J, Rodríguez de la Vega RC, Lopez-Villavicencio M, Gouzy J, Sallet E, Debuchy R, Dupont J, Branca A and Giraud T. 2015. Adaptive horizontal gene transfers between multiple cheese-associated fungi. Current Biology 19, 2562–2569. doi: 10.1016/j.cub.2015.08.025 [4] Novo M, Bigey F, Beyne E, Galeote V, Gavory F, Mallet S, Cambon B, Legras JL, Wincker P, Casaregola S and Dequin S. 2009. Eukaryote-to-eukaryote gene transfer events revealed by the genome sequence of the wine yeast Saccharomyces cerevisiae EC1118. Proceeding of the National Academy of Science USA, 106, 16333–16338. doi: 10.1073/pnas.0904673106 [5] Naranjo-Ortíz MA, Brock M, Brunke S, Hube B, Marcet-Houben M, Gabaldón T. 2016. Widespread inter- and intra-domain horizontal gene transfer of d-amino acid metabolism enzymes in Eukaryotes. Frontiers in Microbiology 7, 2001. doi: 10.3389/fmicb.2016.02001 [6] Alexander WG, Wisecaver JH, Rokas A, Hittinger CT. 2016. Horizontally acquired genes in early-diverging pathogenic fungi enable the use of host nucleosides and nucleotides. Proceeding of the National Academy of Science USA. 113, 4116–4121. doi: 10.1073/pnas.1517242113 [7] Marcet-Houben M, Gabaldón T. 2010. Acquisition of prokaryotic genes by fungal genomes. Trends in Genetics. 26, 5–8. doi: 10.1016/j.tig.2009.11.007 | MaxTiC: Fast ranking of a phylogenetic tree by Maximum Time Consistency with lateral gene transfers | Cédric Chauve, Akbar Rafiey, Adrian A. Davin, Celine Scornavacca, Philippe Veber, Bastien Boussau, Gergely J Szöllosi, Vincent Daubin, and Eric Tannier | Lateral gene transfers (LGTs) between ancient species contain information about the relative timing of species diversification. Specifically, the ancestors of a donor species must have existed before the descendants of the recipient species. Hence... | | Bioinformatics & Computational Biology, Evolutionary Dynamics, Genome Evolution, Life History, Molecular Evolution, Phylogenetics / Phylogenomics | Tatiana Giraud | 2017-06-28 13:40:52 | ||
14 Dec 2023

Genetic sex determination in three closely related hydrothermal vent gastropods, including one species with intersex individualsA shared XY sex chromosome system with variable recombination ratesRecommended by Tanja Schwander based on reviews by Hugo Darras, Daniel Jeffries and 1 anonymous reviewerMany species with separate sexes have evolved sex chromosomes, with the sex-limited chromosomes (i.e. the Y or W chromosomes) exhibiting a wide range of genetic divergences from their homologous X or Z chromosomes (Bachtrog et al., 2014). Variable divergences can result from the cessation of recombination between sex chromosomes that occurred at different time points, with the mechanisms of initiation and expansion of recombination suppression along sex chromosomes remaining poorly understood (Charlesworth, 2017). The study by Castel et al (2023) describes the serendipitous discovery of a shared XY sex chromosome system in three closely related hydrothermal vent gastropods. The X and Y chromosomes appear to still recombine but at variable rates across the three species. This variation makes the gastropod system a very promising focus for future research on sex chromosome evolution. An additional intriguing finding is that some females in one of three gastropod species contain male reproductive tissue in their gonads, providing a fascinating case of a mixed or transitory sexual system. Overall, the study by Castel et al (2023) offers the first insights into the reproduction and sex chromosome system of animals living in deep marine vents, which have remained poorly studied and open outstanding research perspectives on these creatures. References Bachtrog, D., J.E.Mank, C.L.Peichel, M.Kirkpatrick, S.P.Otto, T.L. Ashman, M.W.Hahn, J.Kitano, I.Mayrose, R.Ming, et al. 2014.Sex determination: why so many ways of doing it? PLoSBiol. 12:e1001899. https://doi.org/10.1371/journal.pbio.1001899 Charlesworth, D. Young sex chromosomes in plants and animals. 2019. New Phytologist 224: 1095–1107. https://doi.org/10.1111/nph.16002 Castel J, Pradillon F, Cueff V, Leger G, Daguin-Thiébaut C, Ruault S, Mary J, Hourdez S, Jollivet D, and Broquet T 2023. Genetic sex determination in three closely related hydrothermal vent gastropods, including one species with intersex individuals. bioRxiv, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.04.11.536409 | Genetic sex determination in three closely related hydrothermal vent gastropods, including one species with intersex individuals | Castel J, Pradillon F, Cueff V, Leger G, Daguin-Thiébaut C, Ruault S, Mary J, Hourdez S, Jollivet D, and Broquet T | <p style="text-align: justify;">Molluscs have a wide variety of sexual systems and have undergone many transitions from separate sexes to hermaphroditism or vice versa, which is of interest for studying the evolution of sex determination and diffe... | | Population Genetics / Genomics, Reproduction and Sex | Tanja Schwander | 2023-04-14 11:48:25 | ||
05 May 2020
Meta-population structure and the evolutionary transition to multicellularityThe ecology of evolutionary transitions to multicellularityRecommended by Dustin Brisson based on reviews by 2 anonymous reviewersThe evolutionary transition to multicellular life from free-living, single-celled ancestors has occurred independently in multiple lineages [1-5]. This evolutionary transition to cooperative group living can be difficult to explain given the fitness advantages enjoyed by the non-cooperative, single-celled organisms that still numerically dominate life on earth [1,6,7]. Although several hypotheses have been proposed to explain the transition to multicellularity, a common theme is the abatement of the efficacy of natural selection among the single cells during the free-living stage and the promotion of the efficacy of selection among groups of cells during the cooperative stage, an argument reminiscent of those from George Williams’ seminal book [8,9]. The evolution of life cycles appears to be a key step in the transition to multicellularity as it can align fitness advantages of the single-celled 'reproductive' stage with that of the cooperative 'organismal' stage [9-12]. That is, the evolution of life cycles allows natural selection to operate over timescales longer than that of the doubling time of the free-living cells [13]. Despite the importance of this issue, identifying the range of ecological conditions that reduce the importance of natural selection at the single-celled, free-living stage and increase the importance of selection among groups of cooperating cells has not been addressed empirically. References [1] Maynard Smith, J. and Szathmáry, E. (1995). The Major Transitions in Evolution. Oxford, UK: Freeman. p 346. | Meta-population structure and the evolutionary transition to multicellularity | Caroline J Rose, Katrin Hammerschmidt, Yuriy Pichugin and Paul B Rainey | <p>The evolutionary transition to multicellularity has occurred on numerous occasions, but transitions to complex life forms are rare. While the reasons are unclear, relevant factors include the intensity of within- versus between-group selection ... | | Adaptation, Evolutionary Dynamics, Experimental Evolution | Dustin Brisson | 2019-04-04 12:26:36 | ||
16 Nov 2022

Divergence of olfactory receptors associated with the evolution of assortative mating and reproductive isolation in miceTinder in mice: A match made with the sense of smellRecommended by Christelle Fraïsse based on reviews by Angeles de Cara, Ludovic Claude Maisonneuve and 1 anonymous reviewer
Differentiation-based genome scans lie at the core of speciation and adaptation genomics research. Dating back to Lewontin & Krakauer (1973), they have become very popular with the advent of genomics to identify genome regions of enhanced differentiation relative to neutral expectations. These regions may represent genetic barriers between divergent lineages and are key for studying reproductive isolation. However, genome scan methods can generate a high rate of false positives, primarily if the neutral population structure is not accounted for (Bierne et al. 2013). Moreover, interpreting genome scans can be challenging in the context of secondary contacts between diverging lineages (Bierne et al. 2011), because the coupling between different components of reproductive isolation (local adaptation, intrinsic incompatibilities, mating preferences, etc.) can occur readily, thus preventing the causes of differentiation from being determined. Smadja and collaborators (2022) applied a sophisticated genome scan for trait association (BAYPASS, Gautier 2015) to underlie the genetic basis of a polygenetic behaviour: assortative mating in hybridizing mice. My interest in this neat study mainly relies on two reasons. First, the authors used an ingenious geographical setting (replicate pairs of “Choosy” versus “Non-Choosy” populations) with multi-way comparisons to narrow down the list of candidate regions resulting from BAYPASS. The latter corrects for population structure, handles cost-effective pool-seq data and allows for gene-based analyses that aggregate SNP signals within a gene. These features reinforce the set of outlier genes detected; however, not all are expected to be associated with mating preference. The second reason why this study is valuable to me is that Smadja et al. (2022) complemented the population genomic approach with functional predictions to validate the genetic signal. In line with previous behavioural and chemical assays on the proximal mechanisms of mating preferences, they identified multiple olfactory and vomeronasal receptor genes as highly significant candidates. Therefore, combining genomic signals with functional analyses is a clever way to provide insights into the causes of reproductive isolation, especially when multiple barriers are involved. This is typically true for reinforcement (Butlin & Smadja 2018), suspected to occur in these mice because, in that case, assortative mating (a prezygotic barrier) evolves in response to the cost of hybridization (for example, due to hybrid inviability). As advocated by the authors, their study paves the way for future work addressing the genetic basis of reinforcement, a trait of major evolutionary importance for which we lack empirical data. They also make a compelling case using complementary approaches that olfactory and vomeronasal receptors have a central role in mammal speciation.
Bierne N, Welch J, Loire E, Bonhomme F, David P (2011) The coupling hypothesis: why genome scans may fail to map local adaptation genes. Mol Ecol 20: 2044–2072. https://doi.org/10.1111/j.1365-294X.2011.05080.x Bierne N, Roze D, Welch JJ (2013) Pervasive selection or is it…? why are FST outliers sometimes so frequent? Mol Ecol 22: 2061–2064. https://doi.org/10.1111/mec.12241 Butlin RK, Smadja CM (2018) Coupling, Reinforcement, and Speciation. Am Nat 191:155–172. https://doi.org/10.1086/695136 Gautier M (2015) Genome-Wide Scan for Adaptive Divergence and Association with Population-Specific Covariates. Genetics 201:1555–1579. https://doi.org/10.1534/genetics.115.181453 Lewontin RC, Krakauer J (1973) Distribution of gene frequency as a test of the theory of selective neutrality of polymorphisms. Genetics 74: 175–195. https://doi.org/10.1093/genetics/74.1.175 Smadja CM, Loire E, Caminade P, Severac D, Gautier M, Ganem G (2022) Divergence of olfactory receptors associated with the evolution of assortative mating and reproductive isolation in mice. bioRxiv, 2022.07.21.500634, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.07.21.500634 | Divergence of olfactory receptors associated with the evolution of assortative mating and reproductive isolation in mice | Carole M. Smadja, Etienne Loire, Pierre Caminade, Dany Severac, Mathieu Gautier, Guila Ganem | <p>Deciphering the genetic bases of behavioural traits is essential to understanding how they evolve and contribute to adaptation and biological diversification, but it remains a substantial challenge, especially for behavioural traits with polyge... | | Adaptation, Behavior & Social Evolution, Genotype-Phenotype, Speciation | Christelle Fraïsse | 2022-07-25 11:54:52 | ||
20 Dec 2022
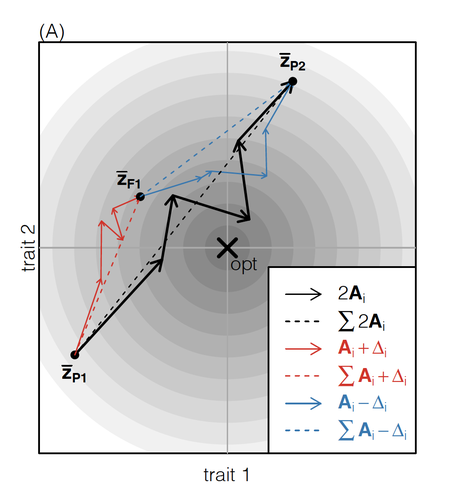
How does the mode of evolutionary divergence affect reproductive isolation?A general model of fitness effects following hybridisationRecommended by Matthew Hartfield based on reviews by Luis-Miguel Chevin and Juan LiStudying the effects of speciation, hybridisation, and evolutionary outcomes following reproduction from divergent populations is a major research area in evolutionary genetics [1]. There are two phenomena that have been the focus of contemporary research. First, a classic concept is the formation of ‘Bateson-Dobzhansky-Muller’ incompatibilities (BDMi) [2–4] that negatively affect hybrid fitness. Here, two diverging populations accumulate mutations over time that are unique to that subpopulation. If they subsequently meet, then these mutations might negatively interact, leading to a loss in fitness or even a complete lack of reproduction. BDMi formation can be complex, involving multiple genes and the fitness changes can depend on the direction of introgression [5]. Second, such secondary contact can instead lead to heterosis, where offspring are fitter than their parental progenitors [6]. Understanding which outcomes are likely to arise require one to know the potential fitness effects of mutations underlying reproductive isolation, to determine whether they are likely to reduce or enhance fitness when hybrids are formed. This is far from an easy task, as it requires one to track mutations at several loci, along with their effects, across a fitness landscape. The work of De Sanctis et al. [7] neatly fills in this knowledge gap, by creating a general mathematical framework for describing the consequences of a cross from two divergent populations. The derivations are based on Fisher’s Geometric Model, which is widely used to quantify selection acting on a general fitness landscape that is affected by several biological traits [8,9], and has previously been used in theoretical studies of hybridisation [10–12]. By doing so, they are able to decompose how divergence at multiple loci affects offspring fitness through both additive and dominance effects. A key result arising from their analyses is demonstrating how offspring fitness can be captured by two main functions. The first one is the ‘net effect of evolutionary change’ that, broadly defined, measures how phenotypically divergent two populations are. The second is the ‘total amount of evolutionary change’, which reflects how many mutations contribute to divergence and the effect sizes captured by each of them. The authors illustrate these measurements using simulations covering different scenarios, demonstrating how different parental states can lead to similar fitness outcomes. They also propose experimental methods to measure the underlying mutational effects. This study neatly demonstrates how complex genetic phenomena underlying hybridisation can be captured using fairly simple mathematical formulae. This powerful approach will thus open the door for future research to investigate hybridisation in more detail, whether it is by expanding on these theoretical models or using the elegant outcomes to quantify fitness effects in experiments.
References 1. Coyne JA, Orr HA. Speciation. Sunderland, Mass: Sinauer Associates; 2004. | How does the mode of evolutionary divergence affect reproductive isolation? | Bianca De Sanctis, Hilde Schneemann, John J. Welch | <p>When divergent populations interbreed, the outcome will be affected by the genomic and phenotypic differences that they have accumulated. In this way, the mode of evolutionary divergence between populations may have predictable consequences for... | | Adaptation, Evolutionary Theory, Hybridization / Introgression, Population Genetics / Genomics, Speciation | Matthew Hartfield | 2022-03-30 14:55:46 |




