
Latest recommendations

| Id | Title▼ | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
25 Sep 2023
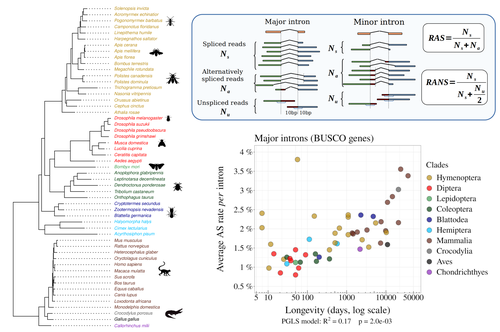
Random genetic drift sets an upper limit on mRNA splicing accuracy in metazoansThe drift barrier hypothesis and the limits to alternative splicing accuracyRecommended by Ignacio Bravo based on reviews by Lars M. Jakt and 2 anonymous reviewersAccurate information flow is central to living systems. The continuity of genomes through generations as well as the reproducible functioning and survival of the individual organisms require a faithful information transfer during replication, transcription and translation. The differential efficiency of natural selection against “mistakes” results in decreasing fidelity rates for replication, transcription and translation. At each level in the information flow chain (replication, transcription, translation), numerous complex molecular systems have evolved and been selected for preventing, identifying and, when possible, correcting or removing such “mistakes” arising during information transfer. However, fidelity cannot be improved ad infinitum. First, because of the limits imposed by the physical nature of the processes of copying and recoding information over different molecular supports: all mechanisms ensuring fidelity during biological information transfer ultimately rely on chemical kinetics and thermodynamics. The more accurate a copying process is, the lower the synthesis rate and the higher the energetic cost of correcting errors. Second, because of the limits imposed by random genetic drift: natural selection cannot effectively act on an allele that contributes with a small differential advantage unless effective population size is large. If s <1/Ne (or s <1/(2Ne) in diploids) the allele frequency in the population is de facto subject to neutral drift processes. In their preprint “Random genetic drift sets an upper limit on mRNA splicing accuracy in metazoans”, Bénitière, Necsulea and Duret explore the validity of this last mentioned “drift barrier” hypothesis for the case study of alternative splicing diversity in eukaryotes (Bénitière et al. 2022). Splicing refers to an ensemble of eukaryotic molecular processes mediated by a large number of proteins and ribonucleoproteins and involving nucleotide sequence recognition, that uses as a molecular substrate a precursor messenger RNA (mRNA), directly transcribed from the DNA, and produces a mature mRNA by removing introns and joining exons (Chow et al. 1977). Alternative splicing refers to the case in which different molecular species of mature mRNAs can be produced, either by cis-splicing processes acting on the same precursor mRNA, e.g. by varying the presence/absence of different exons or by varying the exon-exon boundaries, or by trans-splicing processes, joining exons from different precursor mRNA molecules. The diversity of mRNA molecular species generated by alternative splicing enlarges the molecular phenotypic space that can be generated from the same genotype. In humans, alternative splicing occurs in around 95% of the ca. 20,000 genes, resulting in ca. 100,000 medium-to-high abundance transcripts (Pan et al. 2008). In multicellular organisms, the frequency of alternatively spliced mRNAs varies between tissues and across ontogeny, often in a switch-like pattern (Wang et al. 2008). In the molecular and cell biology community, it is commonly accepted that splice variants contribute with specific functions (Marasco and Kornblihtt 2023) although there exists a discussion around the functional nature of low-frequency splice variants (see for instance the debate between Tress et al. 2017 and Blencowe 2017). The origin, diversity, regulation and evolutionary advantage of alternative splicing constitutes thus a playground of the selectionist-neutralist debate, with one extreme considering that most splice variants are mere “mistakes” of the splicing process (Pickrell et al. 2010), and the other extreme considering that alternative splicing is at the core of complexity in multicellular organisms, as it increases the genome coding potential and allows for a large repertoire of cell types (Chen et al. 2014). In their manuscript, Bénitière, Necsulea and Duret set the cursor towards the neutralist end of the gradient and test the hypothesis of whether the high alternative splice rate in “complex” organisms corresponds to a high rate of splicing “mistakes”, arising from the limit imposed by the drift barrier effect on the power of natural selection to increase accuracy (Bush et al. 2017). In their preprint, the authors convincingly show that in metazoans a fraction of the variation of alternative splicing rate is explained by variation in proxies of population size, so that species with smaller Ne display higher alternative splice rates. They communicate further that abundant splice variants tend to preserve the reading frame more often than low-frequency splice variants, and that the nucleotide splice signals in abundant splice variants display stronger evidence of purifying selection than those in low-frequency splice variants. From all the evidence presented in the manuscript, the authors interpret that “variation in alternative splicing rate is entirely driven by variation in the efficacy of selection against splicing errors”. The authors honestly present some of the limitations of the data used for the analyses, regarding i) the quality of the proxies used for Ne (i.e. body length, longevity and dN/dS ratio); ii) the heterogeneous nature of the RNA sequencing datasets (full organisms, organs or tissues; different life stages, sexes or conditions); and iii) mostly short RNA reads that do not fully span individual introns. Further, data from bacteria do not verify the herein communicated trends, as it has been shown that bacterial species with low population sizes do not display higher transcription error rates (Traverse and Ochman 2016). Finally, it will be extremely interesting to introduce a larger evolutionary perspective on alternative splicing rates encompassing unicellular eukaryotes, in which an intriguing interplay between alternative splicing and gene duplication has been communicated (Hurtig et al. 2020). The manuscript from Bénitière, Necsulea and Duret makes a significant advance to our understanding of the diversity, the origin and the physiology of post-transcriptional and post-translational mechanisms by emphasising the fundamental role of non-adaptive evolutionary processes and the upper limits to splicing accuracy set by genetic drift. References Bénitière F, Necsulea A, Duret L. 2023. Random genetic drift sets an upper limit on mRNA splicing accuracy in metazoans. bioRxiv, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.12.09.519597 Blencowe BJ. 2017. The Relationship between Alternative Splicing and Proteomic Complexity. Trends Biochem Sci 42:407–408. https://doi.org/10.1016/j.tibs.2017.04.001 Bush SJ, Chen L, Tovar-Corona JM, Urrutia AO. 2017. Alternative splicing and the evolution of phenotypic novelty. Philos Trans R Soc Lond B Biol Sci 372:20150474. https://doi.org/10.1098/rstb.2015.0474 Chen L, Bush SJ, Tovar-Corona JM, Castillo-Morales A, Urrutia AO. 2014. Correcting for differential transcript coverage reveals a strong relationship between alternative splicing and organism complexity. Mol Biol Evol 31:1402–1413. https://doi.org/10.1093/molbev/msu083 Chow LT, Gelinas RE, Broker TR, Roberts RJ. 1977. An amazing sequence arrangement at the 5’ ends of adenovirus 2 messenger RNA. Cell 12:1–8. https://doi.org/10.1016/0092-8674(77)90180-5 Hurtig JE, Kim M, Orlando-Coronel LJ, Ewan J, Foreman M, Notice L-A, Steiger MA, van Hoof A. 2020. Origin, conservation, and loss of alternative splicing events that diversify the proteome in Saccharomycotina budding yeasts. RNA 26:1464–1480. https://doi.org/10.1261/rna.075655.120 Marasco LE, Kornblihtt AR. 2023. The physiology of alternative splicing. Nat Rev Mol Cell Biol 24:242–254. https://doi.org/10.1038/s41580-022-00545-z Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. 2008. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40:1413–1415. https://doi.org/10.1038/ng.259 Pickrell JK, Pai AA, Gilad Y, Pritchard JK. 2010. Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet 6:e1001236. https://doi.org/10.1371/journal.pgen.1001236 Traverse CC, Ochman H. 2016. Conserved rates and patterns of transcription errors across bacterial growth states and lifestyles. Proc Natl Acad Sci U S A 113:3311–3316. https://doi.org/10.1073/pnas.1525329113 Tress ML, Abascal F, Valencia A. 2017. Alternative Splicing May Not Be the Key to Proteome Complexity. Trends Biochem Sci 42:98–110. https://doi.org/10.1016/j.tibs.2016.08.008 Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456:470–476. https://doi.org/10.1038/nature07509 | Random genetic drift sets an upper limit on mRNA splicing accuracy in metazoans | Florian Benitiere, Anamaria Necsulea, Laurent Duret | <p style="text-align: justify;">Most eukaryotic genes undergo alternative splicing (AS), but the overall functional significance of this process remains a controversial issue. It has been noticed that the complexity of organisms (assayed by the nu... | | Bioinformatics & Computational Biology, Genome Evolution, Molecular Evolution, Population Genetics / Genomics | Ignacio Bravo | Anonymous | 2022-12-12 14:00:01 | |
11 Dec 2020

Quantifying transmission dynamics of acute hepatitis C virus infections in a heterogeneous population using sequence dataPhylodynamics of hepatitis C virus reveals transmission dynamics within and between risk groups in LyonRecommended by David Rasmussen based on reviews by Chris Wymant and Louis DuPlessisGenomic epidemiology seeks to better understand the transmission dynamics of infectious pathogens using molecular sequence data. Phylodynamic methods have given genomic epidemiology new power to track the transmission dynamics of pathogens by combining phylogenetic analyses with epidemiological modeling. In recent year, applications of phylodynamics to chronic viral infections such as HIV and hepatitis C virus (HVC) have provided some of the best examples of how phylodynamic inference can provide valuable insights into transmission dynamics within and between different subpopulations or risk groups, allowing for more targeted interventions. References [1] Rasmussen, D. A., Volz, E. M., and Koelle, K. (2014). Phylodynamic inference for structured epidemiological models. PLoS Comput Biol, 10(4), e1003570. doi: https://doi.org/10.1371/journal.pcbi.1003570 | Quantifying transmission dynamics of acute hepatitis C virus infections in a heterogeneous population using sequence data | Gonche Danesh, Victor Virlogeux, Christophe Ramière, Caroline Charre, Laurent Cotte, Samuel Alizon | <p>Opioid substitution and syringes exchange programs have drastically reduced hepatitis C virus (HCV) spread in France but HCV sexual transmission in men having sex with men (MSM) has recently arisen as a significant public health concern. The fa... | | Evolutionary Epidemiology, Phylogenetics / Phylogenomics | David Rasmussen | 2019-07-11 13:37:23 | ||
05 Jan 2023

Promoting extinction or minimizing growth? The impact of treatment on trait trajectories in evolving populationsTrait trajectories in evolving populations: insights from mathematical modelsRecommended by Dominik Wodarz based on reviews by Rob Noble and 3 anonymous reviewersThe evolution of cells within organisms can be an important determinant of disease. This is especially clear in the emergence of tumors and cancers from the underlying healthy tissue. In the healthy state, homeostasis is maintained through complex regulatory processes that ensure a relatively constant population size of cells, which is required for tissue function. Tumor cells escape this homeostasis, resulting in uncontrolled growth and consequent disease. Disease progression is driven by further evolutionary processes within the tumor, and so is the response of tumors to therapies. Therefore, evolutionary biology is an important component required for a better understanding of carcinogenesis and the treatment of cancers. In particular, evolutionary theory helps define the principles of mutant evolution and thus to obtain a clearer picture of the determinants of tumor emergence and therapy responses. The study by Raatz and Traulsen [1] makes an important contribution in this respect. They use mathematical and computational models to investigate trait evolution in the context of evolutionary rescue, motivated by the dynamics of cancer, and also bacterial infections. This study views the establishment of tumors as cell dynamics in harsh environments, where the population is prone to extinction unless mutants emerge that increase evolutionary fitness, allowing them to expand (evolutionary rescue). The core processes of the model include growth, death, and mutations. Random mutations are assumed to give rise to cell lineages with different trait combinations, where the birth and death rates of cells can change. The resulting evolutionary trajectories are investigated in the models, and interesting new results were obtained. For example, the turnover of the population was identified as an important determinant of trait evolution. Turnover is defined as the balance between birth and death, with large rates corresponding to fast turnover and small rates to slow turnover. It was found that for fast cell turnover, a given adaptive step in the trait space results in a smaller increase in survival probability than for cell populations with slower turnover. In other words, evolutionary rescue is more difficult to achieve for fast compared to slow turnover populations. While more mutants can be produced for faster cell turnover rates, the analysis showed that this is not sufficient to overcome the barrier to the evolutionary rescue. This result implies that aggressive tumors with fast cell birth and death rates are less likely to persist and progress than tumors with lower turnover rates. This work emphasizes the importance of measuring the turnover rate in different tumors to advance our understanding of the determinants of tumor initiation and progression. The authors discuss that the well-documented heterogeneity in tumors likely also applies to cellular turnover. If a tumor consists of sub-populations with faster and slower turnover, it is possible that a slower turnover cell clone (e.g. characterized by a degree of dormancy) would enjoy a selective advantage. Another source of heterogeneity in turnover could be given by the hierarchical organization of tumors. Similar to the underlying healthy tissue, many tumors are thought to be maintained by a population of cancer stem cells, while the tumor bulk is made up of more differentiated cells. Tissue stem cells tend to be characterized by a lower turnover than progenitor or transit-amplifying cells. Depending on the assumptions about the self-renewal capacity of these different cell populations, the potential for evolutionary rescue could be different depending on the cell compartment in which the mutant emerges. This might be interesting to explore in the future. There are also implications for treatment. Two types of treatment were investigated: density-affecting treatments in which the density of cells is reduced without altering their trait parameters, and trait-affecting treatments in which the birth and/or death rates are altered. Both types of treatment were found to change the trajectories of trait adaptation, which has potentially important practical implications. Interestingly, it was found that competitive release during treatment can result in situations where after treatment cessation, the non-extinct populations recover to reach sizes that were higher than in the absence of treatment. This points towards the potential of adaptive therapy approaches, where sensitive cells are maintained to some extent to suppress resistant clones [2] competitively. In this context, it is interesting that the success of such approaches might also depend on the turnover of the tumor cell population, as shown by a recent mathematical modeling study [3]. In particular, it was found that adaptive therapy is less likely to work for slow compared to fast turnover tumors. Yet, the current study by Raatz and Traulsen [1] suggests that tumors are more likely to evolve in a slow turnover setting. While there is strong relevance of this analysis for tumor evolution, the results generated in this study have more general relevance. Besides tumors, the paper discusses applications to bacterial disease dynamics in some detail, which is also interesting to compare and contrast to evolutionary processes in cancer. Overall, this study provides insights into the dynamics of evolutionary rescue that represent valuable additions to evolutionary theory. References [1] Raatz M, Traulsen A (2023) Promoting extinction or minimizing growth? The impact of treatment on trait trajectories in evolving populations. bioRxiv, 2022.06.17.496570, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.06.17.496570 [2] Gatenby RA, Silva AS, Gillies RJ, Frieden BR (2009) Adaptive Therapy. Cancer Research, 69, 4894–4903. https://doi.org/10.1158/0008-5472.CAN-08-3658 [3] Strobl MAR, West J, Viossat Y, Damaghi M, Robertson-Tessi M, Brown JS, Gatenby RA, Maini PK, Anderson ARA (2021) Turnover Modulates the Need for a Cost of Resistance in Adaptive Therapy. Cancer Research, 81, 1135–1147. https://doi.org/10.1158/0008-5472.CAN-20-0806 | Promoting extinction or minimizing growth? The impact of treatment on trait trajectories in evolving populations | Michael Raatz, Arne Traulsen | <p style="text-align: justify;">When cancers or bacterial infections establish, small populations of cells have to free themselves from homoeostatic regulations that prevent their expansion. Trait evolution allows these populations to evade this r... | | Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Theory | Dominik Wodarz | 2022-06-18 08:44:37 | ||
10 Jan 2020

Probabilities of tree topologies with temporal constraints and diversification shiftsFitting diversification models on undated or partially dated treesRecommended by Nicolas Lartillot based on reviews by Amaury Lambert, Dominik Schrempf and 1 anonymous reviewerPhylogenetic trees can be used to extract information about the process of diversification that has generated them. The most common approach to conduct this inference is to rely on a likelihood, defined here as the probability of generating a dated tree T given a diversification model (e.g. a birth-death model), and then use standard maximum likelihood. This idea has been explored extensively in the context of the so-called diversification studies, with many variants for the models and for the questions being asked (diversification rates shifting at certain time points or in the ancestors of particular subclades, trait-dependent diversification rates, etc). References [1] Didier, G. (2020) Probabilities of tree topologies with temporal constraints and diversification shifts. bioRxiv, 376756, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/376756 | Probabilities of tree topologies with temporal constraints and diversification shifts | Gilles Didier | <p>Dating the tree of life is a task far more complicated than only determining the evolutionary relationships between species. It is therefore of interest to develop approaches apt to deal with undated phylogenetic trees. The main result of this ... | | Bioinformatics & Computational Biology, Macroevolution | Nicolas Lartillot | 2019-01-30 11:28:58 | ||
28 Feb 2023
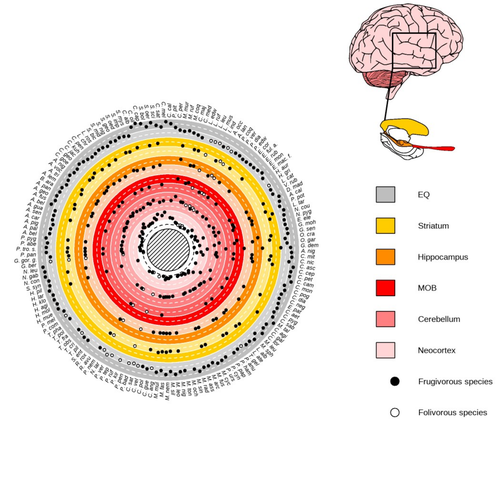
Primate sympatry shapes the evolution of their brain architectureMacroevolutionary drivers of brain evolution in primatesRecommended by Fabien Condamine based on reviews by Paula Gonzalez, Orlin Todorov and 3 anonymous reviewersStudying the evolution of animal cognition is challenging because many environmental and species-related factors can be intertwined, which is further complicated when looking at deep-time evolution. Previous knowledge has emphasized the role of intraspecific interactions in affecting the socio-ecological environment shaping cognition. However, much less is known about such an effect at the interspecific level. Yet, the coexistence of different species in the same geographic area at a given time (sympatry) can impact the evolutionary history of species through character displacement due to biotic interactions. Trait evolution has been observed and tested with morphological external traits but more rarely with brain evolution. Compared to most species’ traits, brain evolution is even more delicate to assess since specific brain regions can be involved in different functions, may they be individual-based and social-based information processing. In a very original and thoroughly executed study, Robira & Perez-Lamarque (2023) addressed the question: How does the co-occurrence of congeneric species shape brain evolution and influence species diversification? By considering brain size as a proxy for cognition, they evaluated whether species sympatry impacted the evolution of cognition in frugivorous primates. Fruit resources are hard to find, not continuous through time, heterogeneously distributed across space, but can be predictable. Hence, cognition considerably shapes the foraging strategy and competition for food access can be fierce. Over long timescales, it remains unclear whether brain size and the pace of species diversification are linked in the context of sympatry, and if so how. Recent studies have found that larger brain sizes can be associated with higher diversification rates in birds (Sayol et al. 2019). Similarly, Robira & Perez-Lamarque (2023) thus wondered if the evolution of brain size in primates impacted their dynamic of species diversification, which has been suggested (Melchionna et al. 2020) but not tested. Prior to anything, Robira & Perez-Lamarque (2023) had to retrace the evolutionary history of sympatry between frugivorous primate lineages through time using the primate tree of life, species’ extant distribution, and process-based models to estimate ancestral range evolution. To infer the effect of species sympatry on the evolution of cognition in frugivorous primates, the authors evaluated the support for phylogenetic models of brain size evolution accounting or not for species sympatry and investigated the directionality of the selection induced by sympatry on brain size evolution. Finally, to better understand the impact of cognition and interactions between primates on their evolutionary success, they tested for correlations between brain size or species’ sympatry and species diversification. Robira & Perez-Lamarque (2023) found that the evolution of the whole brain or brain regions used in immediate information processing was best fitted with models not considering sympatry. By contrast, models considering species sympatry best predicted the evolution of brain regions related to long-term memory of interactions with the socio-ecological environment, with a decrease in their size along with stronger sympatry. Specifically, they found that sympatry was associated with a decrease in the relative size of the hippocampus and striatum, but had no significant effect on the neocortex, cerebellum, or overall brain size. The hippocampus is a brain region that plays a crucial role in processing and memorizing spatiotemporal information, which is relevant for frugivorous primates in their foraging behavior. The study suggests that competition between sympatric species for limited food resources may lead to a more complex and unpredictable food distribution, which may in turn render cognitive foraging not advantageous and result in a selection for smaller brain regions involved in foraging. Niche partitioning and dietary specialization in sympatry may also impact cognitive abilities, with more specialized diets requiring lower cognitive abilities and smaller brain region sizes. On the other hand, the absence of an effect of sympatry on brain regions involved in immediate sensory information processing, such as the cerebellum and neocortex, suggests that foragers do not exploit cues left out by sympatric heterospecific species, or they may discard environmental cues in favor of social cues. This is a remarkable study that highlights the importance of considering the impact of ecological factors, such as sympatry, on the evolution of specific brain regions involved in cognitive processes, and the potential trade-offs in brain region size due to niche partitioning and dietary specialization in sympatry. Further research is needed to explore the mechanisms behind these effects and to test for the possible role of social cues in the evolution of brain regions. This study provides insights into the selective pressures that shape brain evolution in primates. References Melchionna M, Mondanaro A, Serio C, Castiglione S, Di Febbraro M, Rook L, Diniz-Filho JAF, Manzi G, Profico A, Sansalone G, Raia P (2020) Macroevolutionary trends of brain mass in Primates. Biological Journal of the Linnean Society, 129, 14–25. https://doi.org/10.1093/biolinnean/blz161 Robira B, Perez-Lamarque B (2023) Primate sympatry shapes the evolution of their brain architecture. bioRxiv, 2022.05.09.490912, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.05.09.490912 Sayol F, Lapiedra O, Ducatez S, Sol D (2019) Larger brains spur species diversification in birds. Evolution, 73, 2085–2093. https://doi.org/10.1111/evo.13811 | Primate sympatry shapes the evolution of their brain architecture | Benjamin Robira, Benoit Perez-Lamarque | <p style="text-align: justify;">The main hypotheses on the evolution of animal cognition emphasise the role of conspecifics in affecting the socio-ecological environment shaping cognition. Yet, space is often simultaneously occupied by multiple sp... | | Behavior & Social Evolution, Bioinformatics & Computational Biology, Evolutionary Ecology, Macroevolution, Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Fabien Condamine | 2022-05-10 13:43:02 | ||
13 Dec 2016
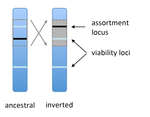
POSTPRINT
Prezygotic isolation, mating preferences, and the evolution of chromosomal inversionsThe spread of chromosomal inversions as a mechanism for reinforcementRecommended by Denis Roze and Thomas Broquet
Several examples of chromosomal inversions carrying genes affecting mate choice have been reported from various organisms. Furthermore, inversions are also frequently involved in genetic isolation between populations or species. Past work has shown that inversions can spread when they capture not only some loci involved in mate choice but also loci involved in incompatibilities between hybridizing populations [1]. In this new paper [2], the authors derive analytical approximations for the selection coefficient associated with an inversion suppressing recombination between a locus involved in mate choice and one (or several) locus involved in Dobzhansky-Muller incompatibilities. Two mechanisms for mate choice are considered: assortative mating based on the allele present at a single locus, or a trait-preference model where one locus codes for the trait and another for the preference. The results show that such an inversion is generally favoured, the selective advantage associated with the inversion being strongest when hybridization is sufficiently frequent. Assuming pairwise epistatic interactions between loci involved in incompatibilities, selection for the inversion increases approximately linearly with the number of such loci captured by the inversion. This paper is a good read for several reasons. First, it presents the problem clearly (e.g. the introduction provides a clear and concise presentation of the issue and past work) and its crystal-clear writing facilitates the reader's understanding of theoretical approaches and results. Second, the analysis is competently done and adds to previous work by showing that very general conditions are expected to be favourable to the spread of the type of inversion considered here. And third, it provides food for thought about the role of inversions in the origin or the reinforcement of divergence between nascent species. One result of this work is that an inversion linked to pre-zygotic isolation "is favoured so long as there is viability selection against recombinant genotypes", suggesting that genetic incompatibilities must have evolved first and that inversions capturing mating preference loci may then enhance pre-existing reproductive isolation. However, the results also show that inversions are more likely to be favoured in hybridizing populations among which gene flow is still high, rather than in more strongly isolated populations. This matches the observation that inversions are more frequently observed between sympatric species than between allopatric ones. References [1] Trickett AJ, Butlin RK. 1994. Recombination Suppressors and the Evolution of New Species. Heredity 73:339-345. doi: 10.1038/hdy.1994.180 [2] Dagilis AJ, Kirkpatrick M. 2016. Prezygotic isolation, mating preferences, and the evolution of chromosomal inversions. Evolution 70: 1465–1472. doi: 10.1111/evo.12954 | Prezygotic isolation, mating preferences, and the evolution of chromosomal inversions | Dagilis AJ, Kirkpatrick M | Chromosomal inversions are frequently implicated in isolating species. Models have shown how inversions can evolve in the context of postmating isolation. Inversions are also frequently associated with mating preferences, a topic that has not been... | | Adaptation, Evolutionary Theory, Genome Evolution, Hybridization / Introgression, Population Genetics / Genomics, Speciation | Denis Roze | 2016-12-13 22:11:54 | ||
26 Oct 2020
Power and limits of selection genome scans on temporal data from a selfing populationDetecting loci under natural selection from temporal genomic data of selfing populationsRecommended by Matteo Fumagalli based on reviews by Christian Huber and 2 anonymous reviewers
The observed levels of genomic diversity in contemporary populations are the result of changes imposed by several evolutionary processes. Among them, natural selection is known to dramatically shape the genetic diversity of loci associated with phenotypes which affect the fitness of carriers. As such, many efforts have been dedicated towards developing methods to detect signatures of natural selection from genomes of contemporary samples [1]. References [1] Stern AJ, Nielsen R (2019) Detecting Natural Selection. In: Handbook of Statistical Genomics , pp. 397–40. John Wiley and Sons, Ltd. https://doi.org/10.1002/9781119487845.ch14 | Power and limits of selection genome scans on temporal data from a selfing population | Miguel Navascués, Arnaud Becheler, Laurène Gay, Joëlle Ronfort, Karine Loridon, Renaud Vitalis | <p>Tracking genetic changes of populations through time allows a more direct study of the evolutionary processes acting on the population than a single contemporary sample. Several statistical methods have been developed to characterize the demogr... | | Adaptation, Bioinformatics & Computational Biology, Population Genetics / Genomics, Reproduction and Sex | Matteo Fumagalli | 2020-05-08 10:34:31 | ||
14 May 2020

Potential adaptive divergence between subspecies and populations of snapdragon plants inferred from QST – FST comparisonsFrom populations to subspecies… to species? Contrasting patterns of local adaptation in closely-related taxa and their potential contribution to species divergenceRecommended by Emmanuelle Porcher based on reviews by Sophie Karrenberg, Santiago C. Gonzalez-Martinez and 1 anonymous reviewerElevation gradients are convenient and widely used natural setups to study local adaptation, particularly in these times of rapid climate change [e.g. 1]. Marin and her collaborators [2] did not follow the mainstream, however. Instead of tackling adaptation to climate change, they used elevation gradients to address another crucial evolutionary question [3]: could adaptation to altitude lead to ecological speciation, i.e. reproductive isolation between populations in spite of gene flow? More specifically, they examined how much local adaptation to environmental variation differed among closely-related, recently diverged subspecies. They studied several populations of two subspecies of snapdragon (Antirrhinum majus), with adjacent geographical distributions. Using common garden experiments and the classical, but still useful, QST-FST comparison, they demonstrate contrasting patterns of local adaptation to altitude between the two subspecies, with several traits under divergent selection in A. majus striatum but none in A. majus pseudomajus. These differences in local adaptation may contribute to species divergence, and open many stimulating questions on the underlying mechanisms, such as the identity of environmental drivers or contribution of reproductive isolation involving flower color polymorphism. References [1] Anderson, J. T., and Wadgymar, S. M. (2020). Climate change disrupts local adaptation and favours upslope migration. Ecology letters, 23(1), 181-192. doi: 10.1111/ele.13427 | Potential adaptive divergence between subspecies and populations of snapdragon plants inferred from QST – FST comparisons | Sara Marin, Anaïs Gibert, Juliette Archambeau, Vincent Bonhomme, Mylène Lascoste and Benoit Pujol | <p>Phenotypic divergence among natural populations can be explained by natural selection or by neutral processes such as drift. Many examples in the literature compare putatively neutral (FST) and quantitative genetic (QST) differentiation in mult... | | Adaptation, Evolutionary Ecology, Genotype-Phenotype, Morphological Evolution, Quantitative Genetics | Emmanuelle Porcher | 2018-08-05 15:34:30 | ||
10 Jul 2019
Population genomics supports clonal reproduction and multiple gains and losses of parasitic abilities in the most devastating nematode plant pestThe scandalous pestRecommended by Nicolas Galtier based on reviews by 2 anonymous reviewersKoutsovoulos et al. [1] have generated and analysed the first population genomic dataset in root-knot nematode Meloidogyne incognita. Why is this interesting? For two major reasons. First, M. incognita has been documented to be apomictic, i.e., to lack any form of sex. This is a trait of major evolutionary importance, with implications on species adaptive potential. The study of genome evolution in asexuals is fascinating and has the potential to inform on the forces governing the evolution of sex and recombination. Even small amounts of sex, however, are sufficient to restore most of the population genetic properties of true sexuals [2]. Because rare events of sex can remain undetected in the field, to confirm asexuality in M. incognita using genomic data is an important step. The second reason why M. incognita is of interest is that this nematode is one of the most harmful pests currently living on earth. M. incognita feeds on the roots of many cultivated plants, including tomato, bean, and cotton, and has been of major agricultural importance for decades. A number of races were defined based on host specificity. These have played a key role in attempts to control the dynamic of M. incognita populations via crop rotations. Races and management strategies so far lack any genetic basis, hence the second major interest of this study. References [1] Koutsovoulos, G. D., Marques, E., Arguel, M. J., Duret, L., Machado, A. C. Z., Carneiro, R. M. D. G., Kozlowski, D. K., Bailly-Bechet, M., Castagnone-Sereno, P., Albuquerque, E. V., & Danchin, E. G. J. (2019). Population genomics supports clonal reproduction and multiple gains and losses of parasitic abilities in the most devastating nematode plant pest. bioRxiv, 362129, ver. 5, peer-reviewed and recommended by Peer Community in Evolutionary Biology. doi: 10.1101/362129 | Population genomics supports clonal reproduction and multiple gains and losses of parasitic abilities in the most devastating nematode plant pest | Georgios D. Koutsovoulos, Eder Marques, Marie-Jeanne Arguel, Laurent Duret, Andressa C.Z. Machado, Regina M.D.G. Carneiro, Djampa K. Kozlowski, Marc Bailly-Bechet, Philippe Castagnone-Sereno, Erika V.S. Albuquerque, Etienne G.J. Danchin | <p>The most devastating nematodes to worldwide agriculture are the root-knot nematodes with Meloidogyne incognita being the most widely distributed and damaging species. This parasitic and ecological success seem surprising given its supposed obli... | | Adaptation, Bioinformatics & Computational Biology, Evolutionary Ecology, Genome Evolution, Genotype-Phenotype, Molecular Evolution, Phylogenetics / Phylogenomics, Population Genetics / Genomics, Reproduction and Sex | Nicolas Galtier | 2018-08-24 09:02:33 | ||
07 Aug 2023

Pollen-feeding delays reproductive senescence and maintains toxicity of Heliconius eratoImpact of pollen-feeding on egg-laying and cyanogenic glucoside abundance in red postman butterfliesRecommended by Adriana Briscoe based on reviews by Carol Boggs, Caroline Mueller and 1 anonymous reviewerGrowth, development and reproduction in animals are all limited by dietary nutrients. Expansion of an organism’s diet to sources not accessible to closely related species reduces food competition, and eases the constraints of nutrient-limited diets. Adult butterflies are herbivorous insects known to feed primarily on nectar from flowers, which is rich in sugars but poor in amino acids. Only certain species in the genus Heliconius are known to also feed on pollen, which is especially rich in amino acids, and is known to prolong their lives by several months. The ability to digest pollen in Heliconius has been linked to specialized feeding behaviors (Krenn et al. 2009) and extra-oral digestion using enzymes, possibly including duplicated copies of cocoonase (Harpel et al. 2016; Smith et al. 2016 and 2018), a protease used by some moths to digest silk upon eclosion from their cocoons. In this reprint, Pinheiro de Castro and colleagues investigated the impact of artificial and natural diets on egg-laying ability, body weight, and cyanogenic glucoside abundance in adult Heliconius erato butterflies of both sexes. Previous studies (Dunlap-Pianka et al. 1981) in H. charithonia demonstrated that access to dietary pollen led to extended egg-laying ability among adult female butterflies compared to females deprived of pollen, and compared to Dryas iulia females which feed only on nectar. In the current study, Pinheiro de Castro et al. (2023) examine the impact of diet on both young and old H. erato, over a longer period of time than the earlier work, highlighting the importance of extending the time period over which effects are evaluated. In addition to extending egg-laying ability in older females, the authors found that pollen in the diet appeared to maintain older female body weight, presumably because the pollen contained nutrients depleted during egg-laying. The authors then investigated the effects of nutrition on the production of cyanogenic glycoside defenses. Heliconius are aposematic butterflies that sequester cyanide-forming defense chemicals from food plants as larvae or synthesize these compounds de novo. The authors found the abundance of cyanogenic glycosides to be significantly greater in butterflies with access to pollen, but again only in older females. Curiously, field studies of male and female H. charithonia butterflies found that females in the wild collected more pollen than males (Mendoza-Cuenca and Macías-Ordóñez 2005). Taken together, these new findings raise the intriguing possibility that females collect more pollen than males, in part, because pollen has a bigger impact on female survival and reproduction. A small limitation of the study is the use of wing length, rather than body weight, at the zero time point. But the trend is clear in both males and females, and it adds supporting detail to the efficacy of pollen feeding as an unusual strategy for increasing fertility and survival in Heliconius butterflies.
References | Pollen-feeding delays reproductive senescence and maintains toxicity of Heliconius erato | Erika C. Pinheiro de Castro, Josie McPherson, Glennis Jullian, Anniina L. K. Mattila, Søren Bak, Stephen Montgomery, Chris Jiggins | <p>Dietary shifts may act to ease energetic constraints and allow organisms to optimise life-history traits. Heliconius butterflies differ from other nectar-feeders due to their unique ability to digest pollen, which provides a reliable source of ... | | Evolutionary Ecology, Life History | Adriana Briscoe | 2023-02-07 12:59:54 |




