
Latest recommendations

| Id | Title | Authors▲ | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
03 Jun 2018
Cost of resistance: an unreasonably expensive conceptLet’s move beyond costs of resistance!Recommended by Inês Fragata and Claudia Bank based on reviews by Danna Gifford, Helen Alexander and 1 anonymous reviewerThe increase in the prevalence of (antibiotic) resistance has become a major global health concern and is an excellent example of the impact of real-time evolution on human society. This has led to a boom of studies that investigate the mechanisms and factors involved in the evolution of resistance, and to the spread of the concept of "costs of resistance". This concept refers to the relative fitness disadvantage of a drug-resistant genotype compared to a non-resistant reference genotype in the ancestral (untreated) environment. In their paper, Lenormand et al. [1] discuss the history of this concept and highlight its caveats and limitations. The authors address both practical and theoretical problems that arise from the simplistic view of "costly resistance" and argue that they can be prejudicial for antibiotic resistance studies. For a better understanding, they visualize their points of critique by means of Fisher's Geometric model. The authors give an interesting historical overview of how the concept arose and speculate that it emerged (during the 1980s) in an attempt by ecologists to spread awareness that fitness can be environment-dependent, and because of the concept's parallels to trade-offs in life-history evolution. They then identify several problems that arise from the concept, which, besides the conceptual misunderstandings that they can cause, are important to keep in mind when designing experimental studies. The authors highlight and explain the following points: Lenormand et al.'s paper [1] is a timely perspective piece in light of the ever-increasing efforts to understand and tackle resistance evolution [2]. Although some readers may shy away from the rather theoretical presentation of the different points of concern, it will be useful for both theoretical and empirical readers by illustrating the misconceptions that can arise from the concept of the cost of resistance. Ultimately, the main lesson to be learned from this paper may not be to ban the term "cost of resistance" from one's vocabulary, but rather to realize that the successful fight against drug resistance requires more differential information than the measurement of fitness effects in a drug-treated vs. non-treated environment in the lab [3-4]. Specifically, a better integration of the ecological aspects of drug resistance evolution and maintenance is needed [5], and we are far from a general understanding of how environmental factors interact and influence an organism's (absolute and relative) fitness and the effect of resistance mutations. References [1] Lenormand T, Harmand N, Gallet R. 2018. Cost of resistance: an unreasonably expensive concept. bioRxiv 276675, ver. 3 peer-reviewed by Peer Community In Evolutionary Biology. doi: 10.1101/276675 | Cost of resistance: an unreasonably expensive concept | Thomas Lenormand, Noemie Harmand, Romain Gallet | <p>The cost of resistance, or the fitness effect of resistance mutation in absence of the drug, is a very widepsread concept in evolutionary genetics and beyond. It has represented an important addition to the simplistic view that resistance mutat... | | Adaptation, Evolutionary Applications, Evolutionary Ecology, Evolutionary Theory, Experimental Evolution, Genotype-Phenotype, Population Genetics / Genomics | Inês Fragata | 2018-03-09 02:22:07 | ||
25 Mar 2019

The joint evolution of lifespan and self-fertilisationEvolution of selfing & lifespan 2.0Recommended by Thomas Bataillon based on reviews by 2 anonymous reviewersFlowering plants display a staggering diversity of both mating systems and life histories, ranging from almost exclusively selfers to obligate outcrossers, very short-lived annual herbs to super long lived trees. One pervasive pattern that has attracted considerable attention is the tight correlation that is found between mating systems and lifespan [1]. Until recently, theoretical explanations for these patterns have relied on static models exploring the consequences of several non-mutually exclusive important process: levels of inbreeding depression and ability to successfully were center stage. This make sense: successful colonization after long‐distance dispersal is far more likely to happen for self‐compatible than for self‐incompatible individuals in a sexually reproducing species. Furthermore, inbreeding depression (essentially a genetically driven phenomenon) and reproductive insurance are expected to shape the evolution of both mating system and lifespan. References | The joint evolution of lifespan and self-fertilisation | Thomas Lesaffre, Sylvain Billiard | <p>In Angiosperms, there exists a strong association between mating system and lifespan. Most self-fertilising species are short-lived and most predominant or obligate outcrossers are long-lived. This association is generally explained by the infl... | | Evolutionary Theory, Life History, Reproduction and Sex | Thomas Bataillon | 2018-09-19 10:03:51 | ||
24 Aug 2022

Density dependent environments can select for extremes of body sizeA population biological modeling approach for life history and body size evolutionRecommended by Wolf Blanckenhorn based on reviews by Frédéric Guillaume and 2 anonymous reviewersBody size evolution is a central theme in evolutionary biology. Particularly the question of when and how smaller body sizes can evolve continues to interest evolutionary ecologists, because most life history models, and the empirical evidence, document that large body size is favoured by natural and sexual selection in most (even small) organisms and environments at most times. How, then, can such a large range of body size and life history syndromes evolve and coexist in nature? The paper by Coulson et al. lifts this question to the level of the population, a relatively novel approach using so-called integral projection (simulation) models (IPMs) (as opposed to individual-based or game theoretical models). As is well outlined by (anonymous) Reviewer 1, and following earlier papers spearheading this approach in other life history contexts, the authors use the well-known carrying capacity (K) of population biology as the ultimate fitness parameter to be maximized or optimized (rather than body size per se), to ultimately identify factors and conditions promoting the evolution of extreme body sizes in nature. They vary (individual or population) size-structured growth trajectories to observe age and size at maturity, surivorship and fecundity/fertility schedules upon evaluating K (see their Fig. 1). Importantly, trade-offs are introduced via density-dependence, either for adult reproduction or for juvenile survival, in two (of several conceivable) basic scenarios (see their Table 2). All other relevant standard life history variables (see their Table 1) are assumed density-independent, held constant or zero (as e.g. the heritability of body size). The authors obtain evidence for disruptive selection on body size in both scenarios, with small size and a fast life history evolving below a threshold size at maturity (at the lowest K) and large size and a slow life history beyond this threshold (see their Fig. 2). Which strategy wins ultimately depends on the fitness benefits of delaying sexual maturity (at larger size and longer lifespan) at the adult stage relative to the preceeding juvenile mortality costs, in agreement with classic life history theory (Roff 1992, Stearns 1992). The modeling approach can be altered and refined to be applied to other key life history parameters and environments. These results can ultimately explain the evolution of smaller body sizes from large body sizes, or vice versa, and their corresponding life history syndromes, depending on the precise environmental circumstances. All reviewers agreed that the approach taken is technically sound (as far as it could be evaluated), and that the results are interesting and worthy of publication. In a first round of reviews various clarifications of the manuscript were suggested by the reviewers. The new version was substantially changed by the authors in response, to the extent that it now is a quite different but much clearer paper with a clear message palatable for the general reader. The writing is now to the point, the paper's focus becomes clear in the Introduction, Methods & Results are much less technical, the Figures illustrative, and the descriptions and interpretations in the Discussion are easy to follow. In general any reader may of course question the choice and realism of the scenarios and underlying assumptions chosen by the authors for simplicity and clarity, for instance no heritability of body size and no cost of reproduction (other than mortality). But this is always the case in modeling work, and the authors acknowledge and in fact suggest concrete extensions and expansions of their approach in the Discussion. References Coulson T., Felmy A., Potter T., Passoni G., Montgomery R.A., Gaillard J.-M., Hudson P.J., Travis J., Bassar R.D., Tuljapurkar S., Marshall D.J., Clegg S.M. (2022) Density-dependent environments can select for extremes of body size. bioRxiv, 2022.02.17.480952, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.02.17.480952 | Density dependent environments can select for extremes of body size | Tim Coulson, Anja Felmy, Tomos Potter, Gioele Passoni, Robert A Montgomery, Jean-Michel Gaillard, Peter J Hudson, Joseph Travis, Ronald D Bassar, Shripad D Tuljapurkar, Dustin Marshall, Sonya M Clegg | <p>Body size variation is an enigma. We do not understand why species achieve the sizes they do, and this means we also do not understand the circumstances under which gigantism or dwarfism is selected. We develop size-structured integral projecti... | | Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Theory, Life History | Wolf Blanckenhorn | 2022-02-21 07:59:04 | ||
11 Sep 2017

POSTPRINT
Less effective selection leads to larger genomesColonisation of subterranean ecosystems leads to larger genome in waterlouse (Aselloidea)Recommended by Benoit Nabholz and Jochen B. W. WolfThe total amount of DNA utilized to store hereditary information varies immensely among eukaryotic organisms. Single copy genome sizes – disregarding differences due to ploidy - differ by more than three orders of magnitude ranging from a few million nucleotides (Mb) to hundreds of billions (Gb). With the ever-increasing availability of fully sequenced genomes we now know that most of the difference is due either to whole genome duplication or to variation in the abundance of repetitive elements. Regarding repetitive elements, the evolutionary forces underlying the large variation 'allowing' more or less elements in a genome remain largely elusive. A tentative correlation between an organism's complexity (however this may be adequately measured) and genome size, the so called C-value paradox [1], has long been dismissed. Studies testing for selection on secondary phenotypic effects associated with genome size (cell size, metabolic rates, nutrient availability) have yielded mixed results. Nonadaptive theories capitalizing on a role of deleterious insertion-deletion mutations and genetic drift as the main drivers have likewise received mixed support [2-3]. Overall, most evidence was derived from analyses across broad taxonomical scales [4-6]. Lefébure and colleagues [7] take a different approach. They confine their considerations to a homogeneous, restricted taxonomical group, isopod crustaceans of the superfamily Aselloidea. This taxonomic focus allows the authors to circumvent many of the confounding factors such as phylogenetic inertia, life history divergence and mutation rate variation that tend to trouble analyses across broad taxonomic timescales. Another important feature of the chosen system is the evolutionary independent transition of habitat use that has occurred at least 11 times. One group of species inhabits subterranean ecosystems (groundwater), another group thrives on surface water. Populations of the former live in low-energy habitats and are expected to be outnumbered by their surface dwelling relatives. Interestingly – and a precondition for the study - the groundwater species have significantly larger genomes (up to 137%). With this unique set-up, the authors are able to investigate the link between genome size and evolutionary forces related to a proxy of long-term population size by removing many of the confounding factors a priori. Upfront, we learn that the dN/dS ratio is higher in the groundwater species. This may either suggest prevalent positive selection or lower efficacy of purifying selection (relaxed constraint) in the group of species in which population sizes are expected to be low. Using a series of population genetic analyses the authors provide compelling evidence for the latter. Analyses are carefully conducted and include models for estimating the intensity and frequency of purifying and positive selection, the DoS (direction of selection) and α statistic. Next the authors also exclude the possibility that increased dN/dS of the subterranean groundwater species may be due to nonfunctionalization, which may result from the subterranean lifestyle. Overall, these analyses suggest relaxed constraint in smaller populations as the most plausible alternative to explain increased dN/dS ratios. In addition to the efficacy of selection, the authors estimate the timing of the ecological transition under the rationale that the amount of time a species may have been exposed to the subterranean habitat may reflect long term population sizes. To calibrate the 'colonization clock' they apply a neat trick based on the degree of degeneration of the opsin gene (as vision tends to get lost in these habitats). When finally testing which parameters may explain differences in genome size all factors – ecological status, selection efficiency as measured by dN/dS and colonization time - turned out to be significant predictors. Direct estimates of the short term effective population size Ne from polymorphism data, however, did not correlate with genome size. Ruling out the effect of other co-variates such as body size and growth rate the authors conclude that genome size was overall best predicted by long-term population size change upon habitat shift. In that the authors provide convincing evidence that the increase in genome size is linked to a decrease in long-term reduction of selection efficiency of subterranean species. Assuming a bias for insertion mutations over deletion mutations (which is usually the case in eukaryotes) this result is in agreement with the theory of mutational hazard [4-6]. This theory proposed by Michael Lynch postulates that the accumulation of non-functional DNA has a weak deleterious effect that can only be efficiently opposed by natural selection in species with high Ne. In conclusion, Lefébure and colleagues provide novel and welcome evidence supporting a 'neutralist' hypothesis of genome size evolution without the need to invoke an adaptive component. Methodologically, the study cautions against the common use of polymorphism-based estimates of Ne which are often obfuscated by transitory demographic change. Instead, alternative measures of selection efficacy linked to long-term population size may serve as better predictors of genome size. We hope that this study will stimulate additional work testing the link between Ne and genome size variation in other taxonomical groups [8-9]. Using genome sequences instead of the transcriptome approach applied here may concomitantly further our understanding of the molecular mechanisms underlying genome size change. References [1] Thomas, CA Jr. 1971. The genetic organization of chromosomes. Annual Review of Genetics 5: 237–256. doi: 10.1146/annurev.ge.05.120171.001321 [2] Ågren JA, Greiner S, Johnson MTJ, Wright SI. 2015. No evidence that sex and transposable elements drive genome size variation in evening primroses. Evolution 69: 1053–1062. doi: 10.1111/evo.12627 [3] Bast J, Schaefer I, Schwander T, Maraun M, Scheu S, Kraaijeveld K. 2016. No accumulation of transposable elements in asexual arthropods. Molecular Biology and Evolution 33: 697–706. doi: 10.1093/molbev/msv261 [4] Lynch M. 2007. The Origins of Genome Architecture. Sinauer Associates. [5] Lynch M, Bobay LM, Catania F, Gout JF, Rho M. 2011. The repatterning of eukaryotic genomes by random genetic drift. Annual Review of Genomics and Human Genetics 12: 347–366. doi: 10.1146/annurev-genom-082410-101412 [6] Lynch M, Conery JS. 2003. The origins of genome complexity. Science 302: 1401–1404. doi: 10.1126/science.1089370 [7] Lefébure T, Morvan C, Malard F, François C, Konecny-Dupré L, Guéguen L, Weiss-Gayet M, Seguin-Orlando A, Ermini L, Der Sarkissian C, Charrier NP, Eme D, Mermillod-Blondin F, Duret L, Vieira C, Orlando L, and Douady CJ. 2017. Less effective selection leads to larger genomes. Genome Research 27: 1016-1028. doi: 10.1101/gr.212589.116 [8] Lower SS, Johnston JS, Stanger-Hall KF, Hjelmen CE, Hanrahan SJ, Korunes K, Hall D. 2017. Genome size in North American fireflies: Substantial variation likely driven by neutral processes. Genome Biolology and Evolution 9: 1499–1512. doi: 10.1093/gbe/evx097 [9] Sessegolo C, Burlet N, Haudry A. 2016. Strong phylogenetic inertia on genome size and transposable element content among 26 species of flies. Biology Letters 12: 20160407. doi: 10.1098/rsbl.2016.0407 | Less effective selection leads to larger genomes | Tristan Lefébure, Claire Morvan, Florian Malard, Clémentine François, Lara Konecny-Dupré, Laurent Guéguen, Michèle Weiss-Gayet, Andaine Seguin-Orlando, Luca Ermini, Clio Der Sarkissian, N. Pierre Charrier, David Eme, Florian Mermillod-Blondin, Lau... | The evolutionary origin of the striking genome size variations found in eukaryotes remains enigmatic. The effective size of populations, by controlling selection efficacy, is expected to be a key parameter underlying genome size evolution. However... | | Evolutionary Theory, Genome Evolution, Molecular Evolution, Population Genetics / Genomics | Benoit Nabholz | 2017-09-08 09:39:23 | ||
18 Dec 2017
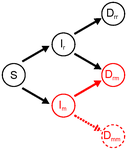
Co-evolution of virulence and immunosuppression in multiple infectionsTwo parasites, virulence and immunosuppression: how does the whole thing evolve?Recommended by Sara Magalhaes based on reviews by 2 anonymous reviewersHow parasite virulence evolves is arguably the most important question in both the applied and fundamental study of host-parasite interactions. Typically, this research area has been progressing through the formalization of the problem via mathematical modelling. This is because the question is a complex one, as virulence is both affected and affects several aspects of the host-parasite interaction. Moreover, the evolution of virulence is a problem in which ecology (epidemiology) and evolution (changes in trait values through time) are tightly intertwined, generating what is now known as eco-evolutionary dynamics. Therefore, intuition is not sufficient to address how virulence may evolve. References [1] Anderson RM and May RM. 1982. Coevolution of hosts and parasites. Parasitology, 1982. 85: 411–426. doi: 10.1017/S0031182000055360 [2] Kamiya T, Mideo N and Alizon S. 2017. Coevolution of virulence and immunosuppression in multiple infections. bioRxiv, ver. 7 peer-reviewed by PCI Evol Biol, 149211. doi: 10.1101/139147 | Co-evolution of virulence and immunosuppression in multiple infections | Tsukushi Kamiya, Nicole Mideo, Samuel Alizon | Many components of the host-parasite interaction have been shown to affect the way virulence, that is parasite induced harm to the host, evolves. However, co-evolution of multiple traits is often neglected. We explore how an immunosuppressive mech... | | Evolutionary Applications, Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Epidemiology, Evolutionary Theory | Sara Magalhaes | 2017-06-13 16:49:45 | ||
31 Mar 2017

POSTPRINT
Human adaptation of Ebola virus during the West African outbreakEbola evolution during the 2013-2016 outbreakRecommended by Sylvain Gandon and Sébastien LionThe Ebola virus (EBOV) epidemic that started in December 2013 resulted in around 28,000 cases and more than 11,000 deaths. Since the emergence of the disease in Zaire in 1976 the virus had produced a number of outbreaks in Africa but until 2013 the reported numbers of human cases had never risen above 500. Could this exceptional epidemic size be due to the spread of a human-adapted form of the virus? The large mutation rate of the virus [1-2] may indeed introduce massive amounts of genetic variation upon which selection may act. Several earlier studies based on the accumulation of genome sequences sampled during the epidemic led to contrasting conclusions. A few studies discussed evidence of positive selection on the glycoprotein that may be linked to phenotypic variations on infectivity and/or immune evasion [3-4]. But the heterogeneity in the transmission of some lineages could also be due to environmental heterogeneity and/or stochasticity. Most studies could not rule out the null hypothesis of the absence of positive selection and human adaptation [1-2 and 5]. In a recent experimental study, Urbanowicz et al. [6] chose a different method to tackle this question. A phylogenetic analysis of genome sequences from viruses sampled in West Africa revealed the existence of two main lineages (one with a narrow geographic distribution in Guinea, and the other with a wider geographic distribution) distinguished by a single amino acid substitution in the glycoprotein of the virus (A82V), and of several sub-lineages characterised by additional substitutions. The authors used this phylogenetic data to generate a panel of mutant pseudoviruses and to test their ability to infect human and fruit bat cells. These experiments revealed that specific amino acid substitutions led to higher infectivity of human cells, including A82V. This increased infectivity on human cells was associated with a decreased infectivity in fruit bat cell cultures. Since fruit bats are likely to be the reservoir of the virus, this paper indicates that human adaptation may have led to a specialization of the virus to a new host. An accompanying paper in the same issue of Cell by Diehl et al. [7] reports results that confirm the trend identified by Urbanowicz et al. [6] and further indicate that the increased infectivity of A82V is specific for primate cells. Diehl et al. [7] also report some evidence for higher virulence of A82V in humans. In other words, the evolution of the virus may have led to higher abilities to infect and to kill its novel host. This work thus confirms the adaptive potential of RNA virus and the ability of Ebola to specialize to a novel host. In this context, the availability of an effective vaccine against the disease is particularly welcome [8]. The study of Urbanowicz et al. [6] is also remarkable because it illustrates the need of experimental approaches for the study of phenotypic variation when inference methods based on phylodynamics fail to extract a clear biological message. The analysis of genomic evolution is still in its infancy and there is a need for new theoretical developments to help detect more rapidly candidate mutations involved in adaptations to new environmental conditions. References [1] Gire, S.K., Goba, A., Andersen, K.G., Sealfon, R.S.G., Park, D.J., Kanneh, L., Jalloh, S., Momoh, M., Fullah, M., Dudas, G., et al. (2014). Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345, 1369–1372. doi: 10.1126/science.1259657 | Human adaptation of Ebola virus during the West African outbreak | Urbanowicz, R.A., McClure, C.P., Sakuntabhai, A., Sall, A.A., Kobinger, G., Müller, M.A., Holmes, E.C., Rey, F.A., Simon-Loriere, E., and Ball, J.K. | The 2013–2016 outbreak of Ebola virus (EBOV) in West Africa was the largest recorded. It began following the cross-species transmission of EBOV from an animal reservoir, most likely bats, into humans, with phylogenetic analysis revealing the co-ci... | | Adaptation, Evolutionary Epidemiology, Genome Evolution, Genotype-Phenotype, Molecular Evolution, Species interactions | Sylvain Gandon | 2017-03-31 14:20:38 | ||
22 Mar 2022
Substantial genetic mixing among sexual and androgenetic lineages within the clam genus CorbiculaStrange reproductive modes and population geneticsRecommended by Chris Jiggins based on reviews by Arnaud Estoup, Simon Henry Martin and 2 anonymous reviewersThere are many organisms that are asexual or have unusual modes of reproduction. One such quasi-sexual reproductive mode is androgenesis, in which the offspring, after fertilization, inherits only the entire paternal nuclear genome. The maternal genome is ditched along the way. One group of organisms which shows this mode of reproduction are clams in the genus Corbicula, some of which are androecious, while others are dioecious and sexual. The study by Vastrade et al. (2022) describes population genetic patterns in these clams, using both nuclear and mitochondrial sequence markers. In contrast to what might be expected for an asexual lineage, there is evidence for significant genetic mixing between populations. In addition, there is high heterozygosity and evidence for polyploidy in some lineages. Overall, the picture is complicated! However, what is clear is that there is far more genetic mixing than expected. One possible mechanism by which this could occur is 'nuclear capture' where there is a mixing of maternal and paternal lineages after fertilization. This can sometimes occur as a result of hybridization between 'species', leading to further mixing of divergent lineages. Thus the group is clearly far from an ancient asexual lineage - recombination and mixing occur with some regularity. The study also analyzed recent invasive populations in Europe and America. These had reduced genetic diversity, but also showed complex patterns of allele sharing suggesting a complex origin of the invasive lineages. In the future, it will be exciting to apply whole genome sequencing approaches to systems such as this. There are challenges in interpreting a handful of sequenced markers especially in a system with polyploidy and considerable complexity, and whole-genome sequencing could clarify some of the outstanding questions, Overall, this paper highlights the complex genetic patterns that can result through unusual reproductive modes, which provides a challenge for the field of population genetics and for the recognition of species boundaries. References Vastrade M, Etoundi E, Bournonville T, Colinet M, Debortoli N, Hedtke SM, Nicolas E, Pigneur L-M, Virgo J, Flot J-F, Marescaux J, Doninck KV (2022) Substantial genetic mixing among sexual and androgenetic lineages within the clam genus Corbicula. bioRxiv, 590836, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/590836 | Substantial genetic mixing among sexual and androgenetic lineages within the clam genus Corbicula | Vastrade M., Etoundi E., Bournonville T., Colinet M., Debortoli N., Hedtke S.M., Nicolas E., Pigneur L.-M., Virgo J., Flot J.-F., Marescaux J. and Van Doninck K. | <p style="text-align: justify;">“Occasional” sexuality occurs when a species combines clonal reproduction and genetic mixing. This strategy is predicted to combine the advantages of both asexuality and sexuality, but its actual consequences on the... | | Evolutionary Ecology, Hybridization / Introgression, Phylogeography & Biogeography | Chris Jiggins | 2019-03-29 15:42:56 | ||
18 Nov 2022
Fitness costs and benefits in response to artificial artesunate selection in PlasmodiumThe importance of understanding fitness costs associated with drug resistance throughout the life cycle of malaria parasitesRecommended by Silvie Huijben based on reviews by Sarah Reece and Marianna SzucsAntimalarial resistance is a major hurdle to malaria eradication efforts. The spread of drug resistance follows basic evolutionary principles, with competitive interactions between resistant and susceptible malaria strains being central to the fitness of resistant parasites. These competitive interactions can be used to design resistance management strategies, whereby a fitness cost of resistant parasites can be exploited through maintaining competitive suppression of the more fit drug-susceptible parasites. This can potentially be achieved using lower drug dosages or lower frequency of drug treatments. This approach has been demonstrated to work empirically in a rodent malaria model [1,2] and has been demonstrated to have clinical success in cancer treatments [3]. However, these resistance management approaches assume a fitness cost of the resistant pathogen, and, in the case of malaria parasites in general, and for artemisinin resistant parasites in particular, there is limited information on the presence of such fitness cost. The best suggestive evidence for the presence of fitness costs comes from the discontinuation of the use of the drug, which, in the case of chloroquine, was followed by a gradual drop in resistance frequency over the following decade [see e.g. 4,5]. However, with artemisinin derivative drugs still in use, alternative ways to study the presence of fitness costs need to be undertaken. References [1] Huijben S, Bell AS, Sim DG, Tomasello D, Mideo N, Day T, et al. 2013. Aggressive chemotherapy and the selection of drug resistant pathogens. PLoS Pathog. 9(9): e1003578. https://doi.org/10.1371/journal.ppat.1003578 [5] Mharakurwa S, Matsena-Zingoni Z, Mudare N, Matimba C, Gara TX, Makuwaza A, et al. 2021. Steep rebound of chloroquine-sensitive Plasmodium falciparum in Zimbabwe. J Infect Dis. 223(2): 306-9. https://doi.org/10.1093/infdis/jiaa368 | Fitness costs and benefits in response to artificial artesunate selection in Plasmodium | Villa M, Berthomieu A, Rivero A | <p style="text-align: justify;">Drug resistance is a major issue in the control of malaria. Mutations linked to drug resistance often target key metabolic pathways and are therefore expected to be associated with biological costs. The spread of dr... | | Evolutionary Applications, Life History | Silvie Huijben | 2022-01-31 13:01:16 | ||
29 Nov 2022

Joint inference of adaptive and demographic history from temporal population genomic dataInference of genome-wide processes using temporal population genomic dataRecommended by Aurelien Tellier based on reviews by Lawrence Uricchio and 2 anonymous reviewersEvolutionary genomics, and population genetics in particular, aim to decipher the respective influence of neutral and selective forces shaping genetic polymorphism in a species/population. This is a much-needed requirement before scanning genome data for footprints of species adaptation to their biotic and abiotic environment (Johri et al. 2022). In general, we would like to quantify the proportion of the genome evolving neutrally and under selective (positive, balancing and negative) pressures (Kern and Hahn 2018, Johri et al. 2021). We thus need to understand patterns of linked selection along the genome, that is how the distribution of genetic polymorphisms is shaped by selected sites and the recombination landscape. The present contribution by Pavinato et al. (2022) provides an additional method in the population genomics toolbox to quantify the extent of linked positive and negative selection using temporal data. The availability of genomics data for model and non-model species has led to improvement of the modeling framework for demography and selection (Johri et al. 2022), but also new inference methods making use of the full genome data based on the Sequential Markovian Coalescent (SMC, Li and Durbin 2011), Approximate Bayesian Computation (ABC, Jay et al. 2019), ABC and machine learning (Pudlo et al. 2016, Raynal et al. 2019) or Deep Learning (Sanchez et al. 2021). These methods are based on one sample in time and the use of the coalescent theory to reconstruct the past (demographic) history. However, it is also possible to obtain for many species temporal data sampled over several time points. For species with short generation time (in experimental evolution or monitored populations), one can sample a population every couple of generations as exemplified with Drosophila melanogaster (Bergland et al. 2010). For species with longer generation times that cannot be easily regularly sampled in time, it becomes possible to sequence available specimens from museums (e.g. Cridland et al. 2018) or ancient DNA samples. Methods using temporal data are based on the classical population genomics assumption that demography (migration, population subdivision, population size changes) leaves a genome-wide signal, while selection leaves a localized signal in the close vicinity of the causal mutation. Several methods do assess the demography of a population (change in effective population size, Ne, in time) using temporal data (e.g. Jorde and Ryman 2007) which can be used to calibrate the detection of loci under strong positive selection (Foll et al. 2014). Recently Buffalo and Coop (2020) used genome-wide covariance between allele frequency changes across time samples (and across replicates) to quantify the effects of linked selection over short timescales. In the present contribution, Pavinato et al. (2022) make use of temporal data to draw the joint estimation of demographic and selective parameters using a simulation-based method (ABC-Random Forests). This study by Pavinato et al. (2022) builds a framework allowing to infer the census size of the population in time (N) separately from the effect of genetic drift, which is determined by change in effective population size (Ne) in time, as well estimates of genome-wide parameters of selection. In a nutshell, the authors use a forward simulator and summarize genome data by genomic windows using classic statistics (nucleotide diversity, Tajima’s D, FST, heterozygosity) between time samples and for each sample. They specifically use the distributions (higher moments) of these statistics among all windows. The authors combine as input for the ABC-RF, vectors of summary statistics, model parameters and five latent variables: Ne, the ratio Ne/N, the number of beneficial mutations under strong selection, the average selection coefficient of strongly selected mutations, and the average substitution load. Indeed, the authors are interested in three different types of selection components: 1) the adaptive potential of a population which is estimated as the population mutation rate of beneficial mutations (θb), 2) the number of mutations under strong selection (irrespective of whether they reached fixation or not), and 3) the overall population fitness which is a function of the genetic load. In other words, the novelty of this method is not to focus on the detection of loci under selection, but to infer key parameters/distributions summarizing the genome-wide signal of demography and (positive and negative) selection. As a proof of principle, the authors then apply their method to a dataset of feral populations of honey bees (Apis mellifera) collected in California across many years and recovered from Museum samples (Cridland et al. 2018). The approach yields estimates of Ne which are on the same order of magnitude of previous estimates in hymenopterans, and the authors discuss why the different populations show various values of Ne and N which can be explained by different history of admixture with wild but also domesticated lineages of bees. This study focuses on quantifying the genome-wide joint footprints of demography, and strong positive and negative selection to determine which proportion of the genome evolves neutrally or not. Further application of this method can be anticipated, for example, to study species with ecological and life-history traits which generate discrepancies between census size and Ne, for example for plants with selfing or seed banking (Sellinger et al. 2020), and for which the genome-wide effect of linked selection is not fully understood. References Johri P, Aquadro CF, Beaumont M, Charlesworth B, Excoffier L, Eyre-Walker A, Keightley PD, Lynch M, McVean G, Payseur BA, Pfeifer SP, Stephan W, Jensen JD (2022) Recommendations for improving statistical inference in population genomics. PLOS Biology, 20, e3001669. https://doi.org/10.1371/journal.pbio.3001669 Kern AD, Hahn MW (2018) The Neutral Theory in Light of Natural Selection. Molecular Biology and Evolution, 35, 1366–1371. https://doi.org/10.1093/molbev/msy092 Johri P, Riall K, Becher H, Excoffier L, Charlesworth B, Jensen JD (2021) The Impact of Purifying and Background Selection on the Inference of Population History: Problems and Prospects. Molecular Biology and Evolution, 38, 2986–3003. https://doi.org/10.1093/molbev/msab050 Pavinato VAC, Mita SD, Marin J-M, Navascués M de (2022) Joint inference of adaptive and demographic history from temporal population genomic data. bioRxiv, 2021.03.12.435133, ver. 6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.03.12.435133 Li H, Durbin R (2011) Inference of human population history from individual whole-genome sequences. Nature, 475, 493–496. https://doi.org/10.1038/nature10231 Jay F, Boitard S, Austerlitz F (2019) An ABC Method for Whole-Genome Sequence Data: Inferring Paleolithic and Neolithic Human Expansions. Molecular Biology and Evolution, 36, 1565–1579. https://doi.org/10.1093/molbev/msz038 Pudlo P, Marin J-M, Estoup A, Cornuet J-M, Gautier M, Robert CP (2016) Reliable ABC model choice via random forests. Bioinformatics, 32, 859–866. https://doi.org/10.1093/bioinformatics/btv684 Raynal L, Marin J-M, Pudlo P, Ribatet M, Robert CP, Estoup A (2019) ABC random forests for Bayesian parameter inference. Bioinformatics, 35, 1720–1728. https://doi.org/10.1093/bioinformatics/bty867 Sanchez T, Cury J, Charpiat G, Jay F (2021) Deep learning for population size history inference: Design, comparison and combination with approximate Bayesian computation. Molecular Ecology Resources, 21, 2645–2660. https://doi.org/10.1111/1755-0998.13224 Bergland AO, Behrman EL, O’Brien KR, Schmidt PS, Petrov DA (2014) Genomic Evidence of Rapid and Stable Adaptive Oscillations over Seasonal Time Scales in Drosophila. PLOS Genetics, 10, e1004775. https://doi.org/10.1371/journal.pgen.1004775 Cridland JM, Ramirez SR, Dean CA, Sciligo A, Tsutsui ND (2018) Genome Sequencing of Museum Specimens Reveals Rapid Changes in the Genetic Composition of Honey Bees in California. Genome Biology and Evolution, 10, 458–472. https://doi.org/10.1093/gbe/evy007 Jorde PE, Ryman N (2007) Unbiased Estimator for Genetic Drift and Effective Population Size. Genetics, 177, 927–935. https://doi.org/10.1534/genetics.107.075481 Foll M, Shim H, Jensen JD (2015) WFABC: a Wright–Fisher ABC-based approach for inferring effective population sizes and selection coefficients from time-sampled data. Molecular Ecology Resources, 15, 87–98. https://doi.org/10.1111/1755-0998.12280 Buffalo V, Coop G (2020) Estimating the genome-wide contribution of selection to temporal allele frequency change. Proceedings of the National Academy of Sciences, 117, 20672–20680. https://doi.org/10.1073/pnas.1919039117 Sellinger TPP, Awad DA, Moest M, Tellier A (2020) Inference of past demography, dormancy and self-fertilization rates from whole genome sequence data. PLOS Genetics, 16, e1008698. https://doi.org/10.1371/journal.pgen.1008698 | Joint inference of adaptive and demographic history from temporal population genomic data | Vitor A. C. Pavinato, Stéphane De Mita, Jean-Michel Marin, Miguel de Navascués | <p style="text-align: justify;">Disentangling the effects of selection and drift is a long-standing problem in population genetics. Simulations show that pervasive selection may bias the inference of demography. Ideally, models for the inference o... | | Adaptation, Population Genetics / Genomics | Aurelien Tellier | 2021-10-20 09:41:26 | ||
11 Oct 2022
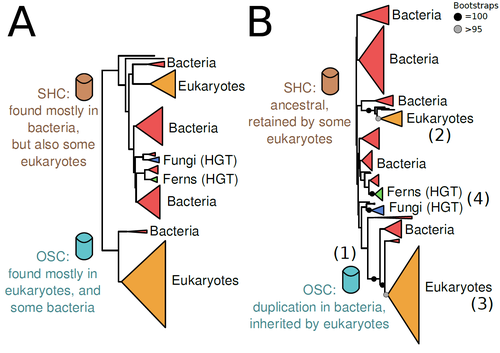
The Eukaryotic Last Common Ancestor Was Bifunctional for Hopanoid and Sterol ProductionGene family analysis suggests new evolutionary scenario for sterol and hopanoid biomarkersRecommended by Iker Irisarri based on reviews by Samuel Abalde, Denis Baurain and Jose Ramon Pardos-BlasSterols and hopanoids are sometimes used as biomarkers to infer the origin of certain groups of organisms. Traditionally, hopanoid-derived products in ancient rocks have been considered to indicate the presence of bacteria, whereas sterol derivatives have been considered to be exclusive to eukaryotes. However, a closer look at the topic reveals a rather complex distribution of either compound in both bacteria and eukaryotes. (1). The known biosynthetic pathways for sterols and hopanoids are similar but diverge at a critical step where two different enzymes are used: squalene-hopene cyclase (SHC) and oxidosqualene cyclase (OSC), the latter requiring oxygen. These two enzymes belong to the same gene family, whose complex evolutionary history is difficult to reconcile with the known species phylogeny. In this study (2), Dr. Warren R. Francis revisits the evolution of this gene family using an extended dataset with a broader taxonomic representation. In contrast to the traditional representation of the tree rooted between SHC and OSC paralogs (i.e., based on function), the author proposes that rooting the tree within bacterial SHCs and assuming a secondary origin of OSC is more parsimonious. This postulates SHC to be the ancestral function –retained in many extant bacteria and some eukaryotes– and OSC to have emerged later within bacteria –currently being mostly present in eukaryotes–. The reconstructed evolutionary history is arguably complex and can only be reconciled with the species' phylogeny by invoking many secondary losses. These losses are considered likely because many extant species acquire sterols and hopanoids by diet and lack one or both enzymes. Some cases of recent horizontal gene transfer are also proposed. In contrast to the dichotomy between bacterial SHCs and eukaryote OSCs, the new proposed scenario suggests that the eukaryote ancestor likely inherited both enzymes from bacteria and thus could be able to synthesize both sterols and hopanoids. Under this hypothesis, not only bacteria but also eukaryotes could be responsible for the hopane found in old rocks. This agrees with eukaryote fossils dating back to more than 1 billion years ago (3). Also, the observed increase of sterane levels in rocks ~600-700 million years old cannot be associated with the origin of eukaryotes, which is a much older event, but could rather reflect changes in atmospheric oxygen levels because oxygen is required for the synthesis of sterols by OSC. References 1. Santana-Molina C, Rivas-Marin E, Rojas AM, Devos DP (2020) Origin and Evolution of Polycyclic Triterpene Synthesis. Molecular Biology and Evolution, 37, 1925–1941. https://doi.org/10.1093/molbev/msaa054 2. Francis WR (2022) The Eukaryotic Last Common Ancestor Was Bifunctional for Hopanoid and Sterol Production. Preprints, 2020040186, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.20944/preprints202004.0186.v5 3. Butterfield NJ (2000) Bangiomorpha pubescens n. gen., n. sp.: implications for the evolution of sex, multicellularity, and the Mesoproterozoic/Neoproterozoic radiation of eukaryotes. Paleobiology, 26, 386–404. https://doi.org/10.1666/0094-8373(2000)026<0386:BPNGNS>2.0.CO;2 | The Eukaryotic Last Common Ancestor Was Bifunctional for Hopanoid and Sterol Production | Warren R Francis | <p>Steroid and hopanoid biomarkers can be found in ancient rocks and may give a glimpse of what life was present at that time. Sterols and hopanoids are produced by two related enzymes, though the evolutionary history of this protein family is com... | | Bioinformatics & Computational Biology, Evolutionary Ecology, Molecular Evolution, Paleontology, Phylogenetics / Phylogenomics | Iker Irisarri | 2021-01-13 16:03:29 |




