
Latest recommendations

| Id | Title | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers▲ | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
05 May 2020
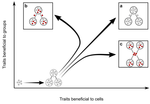
Meta-population structure and the evolutionary transition to multicellularityThe ecology of evolutionary transitions to multicellularityRecommended by Dustin Brisson based on reviews by 2 anonymous reviewersThe evolutionary transition to multicellular life from free-living, single-celled ancestors has occurred independently in multiple lineages [1-5]. This evolutionary transition to cooperative group living can be difficult to explain given the fitness advantages enjoyed by the non-cooperative, single-celled organisms that still numerically dominate life on earth [1,6,7]. Although several hypotheses have been proposed to explain the transition to multicellularity, a common theme is the abatement of the efficacy of natural selection among the single cells during the free-living stage and the promotion of the efficacy of selection among groups of cells during the cooperative stage, an argument reminiscent of those from George Williams’ seminal book [8,9]. The evolution of life cycles appears to be a key step in the transition to multicellularity as it can align fitness advantages of the single-celled 'reproductive' stage with that of the cooperative 'organismal' stage [9-12]. That is, the evolution of life cycles allows natural selection to operate over timescales longer than that of the doubling time of the free-living cells [13]. Despite the importance of this issue, identifying the range of ecological conditions that reduce the importance of natural selection at the single-celled, free-living stage and increase the importance of selection among groups of cooperating cells has not been addressed empirically. References [1] Maynard Smith, J. and Szathmáry, E. (1995). The Major Transitions in Evolution. Oxford, UK: Freeman. p 346. | Meta-population structure and the evolutionary transition to multicellularity | Caroline J Rose, Katrin Hammerschmidt, Yuriy Pichugin and Paul B Rainey | <p>The evolutionary transition to multicellularity has occurred on numerous occasions, but transitions to complex life forms are rare. While the reasons are unclear, relevant factors include the intensity of within- versus between-group selection ... | | Adaptation, Evolutionary Dynamics, Experimental Evolution | Dustin Brisson | 2019-04-04 12:26:36 | ||
13 Sep 2019

Deceptive combined effects of short allele dominance and stuttering: an example with Ixodes scapularis, the main vector of Lyme disease in the U.S.A.New curation method for microsatellite markers improves population genetics analysesRecommended by Aurelien Tellier based on reviews by Eric Petit, Martin Husemann and 2 anonymous reviewersGenetic markers are used for in modern population genetics/genomics to uncover the past neutral and selective history of population and species. Besides Single Nucleotide Polymorphisms (SNPs) obtained from whole genome data, microsatellites (or Short Tandem Repeats, SSR) have been common markers of choice in numerous population genetics studies of non-model species with large sample sizes [1]. Microsatellites can be used to uncover and draw inference of the past population demography (e.g. expansion, decline, bottlenecks…), population split, population structure and gene flow, but also life history traits and modes of reproduction (e.g. [2,3]). These markers are widely used in conservation genetics [4] or to study parasites or disease vectors [5]. Microsatellites do show higher mutation rate than SNPs increasing, on the one hand, the statistical power to infer recent events (for example crop domestication, [2,3]), while, on the other hand, decreasing their statistical power over longer time scales due to homoplasy [6]. References [1] Jarne, P., and Lagoda, P. J. (1996). Microsatellites, from molecules to populations and back. Trends in ecology & evolution, 11(10), 424-429. doi: 10.1016/0169-5347(96)10049-5 | Deceptive combined effects of short allele dominance and stuttering: an example with Ixodes scapularis, the main vector of Lyme disease in the U.S.A. | Thierry De Meeûs, Cynthia T. Chan, John M. Ludwig, Jean I. Tsao, Jaymin Patel, Jigar Bhagatwala, and Lorenza Beati | <p>Null alleles, short allele dominance (SAD), and stuttering increase the perceived relative inbreeding of individuals and subpopulations as measured by Wright’s FIS and FST. Ascertainment bias, due to such amplifying problems are usually caused ... | | Evolutionary Ecology, Other, Population Genetics / Genomics | Aurelien Tellier | 2019-05-02 20:52:08 | ||
09 Dec 2019
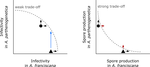
Trait-specific trade-offs prevent niche expansion in two parasitesTrade-offs in fitness components and ecological source-sink dynamics affect host specialisation in two parasites of Artemia shrimpsRecommended by Frédéric Guillaume based on reviews by Anne Duplouy, Seth Barribeau and Cindy Gidoin based on reviews by Anne Duplouy, Seth Barribeau and Cindy Gidoin
Ecological specialisation, especially among parasites infecting a set of host species, is ubiquitous in nature. Host specialisation can be understood as resulting from trade-offs in parasite infectivity, virulence and growth. However, it is not well understood how variation in these trade-offs shapes the overall fitness trade-off a parasite faces when adapting to multiple hosts. For instance, it is not clear whether a strong trade-off in one fitness component may sufficiently constrain the evolution of a generalist parasite despite weak trade-offs in other components. A second mechanism explaining variation in specialisation among species is habitat availability and quality. Rare habitats or habitats that act as ecological sinks will not allow a species to persist and adapt, preventing a generalist phenotype to evolve. Understanding the prevalence of those mechanisms in natural systems is crucial to understand the emergence and maintenance of host specialisation, and biodiversity in general. References [1] Lievens, E.J.P., Michalakis, Y. and Lenormand, T. (2019). Trait-specific trade-offs prevent niche expansion in two parasites. bioRxiv, 621581, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/621581 | Trait-specific trade-offs prevent niche expansion in two parasites | Eva JP Lievens, Yannis Michalakis, Thomas Lenormand | <p>The evolution of host specialization has been studied intensively, yet it is still often difficult to determine why parasites do not evolve broader niches – in particular when the available hosts are closely related and ecologically similar. He... | | Adaptation, Evolutionary Ecology, Evolutionary Epidemiology, Experimental Evolution, Life History, Species interactions | Frédéric Guillaume | 2019-05-13 13:44:34 | ||
28 Aug 2019

Is adaptation limited by mutation? A timescale-dependent effect of genetic diversity on the adaptive substitution rate in animalsTo tinker, evolution needs a supply of spare partsRecommended by Georgii Bazykin based on reviews by Konstantin Popadin, David Enard and 1 anonymous reviewerIs evolution adaptive? Not if there is no variation for natural selection to work with. Theory predicts that how fast a population can adapt to a new environment can be limited by the supply of new mutations coming into it. This supply, in turn, depends on two things: how often mutations occur and in how many individuals. If there are few mutations, or few individuals in whom they can originate, individuals will be mostly identical in their DNA, and natural selection will be impotent. References [1] G, J. A., Visser, M. de, Zeyl, C. W., Gerrish, P. J., Blanchard, J. L., and Lenski, R. E. (1999). Diminishing Returns from Mutation Supply Rate in Asexual Populations. Science, 283(5400), 404–406. doi: 10.1126/science.283.5400.404 | Is adaptation limited by mutation? A timescale-dependent effect of genetic diversity on the adaptive substitution rate in animals | Marjolaine Rousselle, Paul Simion, Marie-Ka Tilak, Emeric Figuet, Benoit Nabholz, Nicolas Galtier | <p>Whether adaptation is limited by the beneficial mutation supply is a long-standing question of evolutionary genetics, which is more generally related to the determination of the adaptive substitution rate and its relationship with the effective... | | Adaptation, Evolutionary Theory, Genome Evolution, Molecular Evolution, Population Genetics / Genomics | Georgii Bazykin | 2019-05-21 09:49:16 | ||
03 May 2020

When does gene flow facilitate evolutionary rescue?Reconciling the upsides and downsides of migration for evolutionary rescueRecommended by Claudia Bank based on reviews by 3 anonymous reviewersThe evolutionary response of populations to changing or novel environments is a topic that unites the interests of evolutionary biologists, ecologists, and biomedical researchers [1]. A prominent phenomenon in this research area is evolutionary rescue, whereby a population that is otherwise doomed to extinction survives due to the spread of new or pre-existing mutations that are beneficial in the new environment. Scenarios of evolutionary rescue require a specific set of parameters: the absolute growth rate has to be negative before the rescue mechanism spreads, upon which the growth rate becomes positive. However, potential examples of its relevance exist (e.g., [2]). From a theoretical point of view, the technical challenge but also the beauty of evolutionary rescue models is that they combine the study of population dynamics (i.e., changes in the size of populations) and population genetics (i.e., changes in the frequencies in the population). Together, the potential relevance of evolutionary rescue in nature and the models' theoretical appeal has resulted in a suite of modeling studies on the subject in recent years. References [1] Bell, G. (2017). Evolutionary Rescue. Annual Review of Ecology, Evolution, and Systematics 48(1), 605-627. doi: 10.1146/annurev-ecolsys-110316-023011 | When does gene flow facilitate evolutionary rescue? | Matteo Tomasini, Stephan Peischl | <p>Experimental and theoretical studies have highlighted the impact of gene flow on the probability of evolutionary rescue in structured habitats. Mathematical modelling and simulations of evolutionary rescue in spatially or otherwise structured p... | | Evolutionary Dynamics, Evolutionary Theory, Population Genetics / Genomics | Claudia Bank | 2019-05-22 11:12:13 | ||
26 Nov 2019
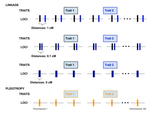
Pleiotropy or linkage? Their relative contributions to the genetic correlation of quantitative traits and detection by multi-trait GWA studiesUnderstanding the effects of linkage and pleiotropy on evolutionary adaptationRecommended by Kathleen Lotterhos based on reviews by Pär Ingvarsson and 1 anonymous reviewerGenetic correlations among traits are ubiquitous in nature. However, we still have a limited understanding of the genetic architecture of trait correlations. Some genetic correlations among traits arise because of pleiotropy - single mutations or genotypes that have effects on multiple traits. Other genetic correlations among traits arise because of linkage among mutations that have independent effects on different traits. Teasing apart the differential effects of pleiotropy and linkage on trait correlations is difficult, because they result in very similar genetic patterns. However, understanding these differential effects gives important insights into how ubiquitous pleiotropy may be in nature. References [1] Chebib, J. and Guillaume, F. (2019). Pleiotropy or linkage? Their relative contributions to the genetic correlation of quantitative traits and detection by multi-trait GWA studies. bioRxiv, 656413, v3 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/656413 | Pleiotropy or linkage? Their relative contributions to the genetic correlation of quantitative traits and detection by multi-trait GWA studies | Jobran Chebib and Frédéric Guillaume | <p>Genetic correlations between traits may cause correlated responses to selection depending on the source of those genetic dependencies. Previous models described the conditions under which genetic correlations were expected to be maintained. Sel... | | Bioinformatics & Computational Biology, Evolutionary Applications, Evolutionary Dynamics, Evolutionary Theory, Genome Evolution, Genotype-Phenotype, Molecular Evolution, Population Genetics / Genomics, Quantitative Genetics | Kathleen Lotterhos | 2019-06-05 13:51:43 | ||
20 Jan 2020

A young age of subspecific divergence in the desert locust Schistocerca gregaria, inferred by ABC Random ForestEstimating recent divergence history: making the most of microsatellite data and Approximate Bayesian Computation approachesRecommended by Takeshi Kawakami and Concetta Burgarella based on reviews by Michael D Greenfield and 2 anonymous reviewersThe present-day distribution of extant species is the result of the interplay between their past population demography (e.g., expansion, contraction, isolation, and migration) and adaptation to the environment. Shedding light on the timing and magnitude of key demographic events helps identify potential drivers of such events and interaction of those drivers, such as life history traits and past episodes of environmental shifts. The understanding of the key factors driving species evolution gives important insights into how the species may respond to changing conditions, which can be particularly relevant for the management of harmful species, such as agricultural pests (e.g. [1]). Meaningful demographic inferences present major challenges. These include formulating evolutionary scenarios fitting species biology and the eco-geographical context and choosing informative molecular markers and accurate quantitative approaches to statistically compare multiple demographic scenarios and estimate the parameters of interest. A further issue comes with result interpretation. Accurately dating the inferred events is far from straightforward since reliable calibration points are necessary to translate the molecular estimates of the evolutionary time into absolute time units (i.e. years). This can be attempted in different ways, such as by using fossil and archaeological records, heterochronous samples (e.g. ancient DNA), and/or mutation rate estimated from independent data (e.g. [2], [3] for review). Nonetheless, most experimental systems rarely meet these conditions, hindering the comprehensive interpretation of results. The contribution of Chapuis et al. [4] addresses these issues to investigate the recent history of the African insect pest Schistocerca gregaria (desert locust). They apply Approximate Bayesian Computation-Random Forest (ABC-RF) approaches to microsatellite markers. Owing to their fast mutation rate microsatellite markers offer at least two advantages: i) suitability for analyzing recently diverged populations, and ii) direct estimate of the germline mutation rate in pedigree samples. The work of Chapuis et al. [4] benefits of both these advantages, since they have estimates of mutation rate and allele size constraints derived from germline mutations in the species [5]. The main aim of the study is to infer the history of divergence of the two subspecies of the desert locust, which have spatially disjoint distribution corresponding to the dry regions of North and West-South Africa. They first use paleo-vegetation maps to formulate hypotheses about changes in species range since the last glacial maximum. Based on them, they generate 12 divergence models. For the selection of the demographic model and parameter estimation, they apply the recently developed ABC-RF approach, a powerful inferential tool that allows optimizing the use of summary statistics information content, among other advantages [6]. Some methodological novelties are also introduced in this work, such as the computation of the error associated with the posterior parameter estimates under the best scenario. The accuracy of timing estimate is assured in two ways: i) by the use of microsatellite markers with known evolutionary dynamics, as underlined above, and ii) by assessing the divergence time threshold above which posterior estimates are likely to be biased by size homoplasy and limits in allele size range [7]. The best-supported model suggests a recent divergence event of the subspecies of S. gregaria (around 2.6 kya) and a reduction of populations size in one of the subspecies (S. g. flaviventris) that colonized the southern distribution area. As such, results did not support the hypothesis that the southward colonization was driven by the expansion of African dry environments associated with the last glacial maximum, as it has been postulated for other arid-adapted species with similar African disjoint distributions [8]. The estimated time of divergence points at a much more recent origin for the two subspecies, during the late Holocene, in a period corresponding to fairly stable arid conditions similar to current ones [9,10]. Although the authors cannot exclude that their microsatellite data bear limited information on older colonization events than the last one, they bring arguments in favour of alternative explanations. The hypothesis privileged does not involve climatic drivers, but the particularly efficient dispersal behaviour of the species, whose individuals are able to fly over long distances (up to thousands of kilometers) under favourable windy conditions. A single long-distance dispersal event by a few individuals would explain the genetic signature of the bottleneck. There is a growing number of studies in phylogeography in arid regions in the Southern hemisphere, but the impact of past climate changes on the species distribution in this region remains understudied relative to the Northern hemisphere [11,12]. The study presented by Chapuis et al. [4] offers several important insights into demographic changes and the evolutionary history of an agriculturally important pest species in Africa, which could also mirror the history of other organisms in the continent. As the authors point out, there are necessarily some uncertainties associated with the models of past ecosystems and climate, especially for Africa. Interestingly, the authors argue that the information on paleo-vegetation turnover was more informative than climatic niche modeling for the purpose of their study since it made them consider a wider range of bio-geographical changes and in turn a wider range of evolutionary scenarios (see discussion in Supplementary Material). Microsatellite markers have been offering a useful tool in population genetics and phylogeography for decades, but their popularity is perhaps being taken over by single nucleotide polymorphism (SNP) genotyping and whole-genome sequencing (WGS) (the peak year of the number of the publication with “microsatellite” is in 2012 according to PubMed). This study reaffirms the usefulness of these classic molecular markers to estimate past demographic events, especially when species- and locus-specific microsatellite mutation features are available and a powerful inferential approach is adopted. Nonetheless, there are still hurdles to overcome, such as the limitations in scenario choice associated with the simulation software used (e.g. not allowing for continuous gene flow in this particular case), which calls for further improvement of simulation tools allowing for more flexible modeling of demographic events and mutation patterns. In sum, this work not only contributes to our understanding of the makeup of the African biodiversity but also offers a useful statistical framework, which can be applied to a wide array of species and molecular markers (microsatellites, SNPs, and WGS). References [1] Lehmann, P. et al. (2018). Complex responses of global insect pests to climate change. bioRxiv, 425488. doi: https://dx.doi.org/10.1101/425488 [2] Donoghue, P. C., & Benton, M. J. (2007). Rocks and clocks: calibrating the Tree of Life using fossils and molecules. Trends in Ecology & Evolution, 22(8), 424-431. doi: https://dx.doi.org/10.1016/j.tree.2007.05.005 [3] Ho, S. Y., Lanfear, R., Bromham, L., Phillips, M. J., Soubrier, J., Rodrigo, A. G., & Cooper, A. (2011). Time‐dependent rates of molecular evolution. Molecular ecology, 20(15), 3087-3101. doi: https://dx.doi.org/10.1111/j.1365-294X.2011.05178.x [4] Chapuis, M.-P., Raynal, L., Plantamp, C., Meynard, C. N., Blondin, L., Marin, J.-M. and Estoup, A. (2020). A young age of subspecific divergence in the desert locust Schistocerca gregaria, inferred by ABC Random Forest. bioRxiv, 671867, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: https://dx.doi.org/10.1101/671867 5] Chapuis, M.-P., Plantamp, C., Streiff, R., Blondin, L., & Piou, C. (2015). Microsatellite evolutionary rate and pattern in Schistocerca gregaria inferred from direct observation of germline mutations. Molecular ecology, 24(24), 6107-6119. doi: https://dx.doi.org/10.1111/mec.13465 [6] Raynal, L., Marin, J. M., Pudlo, P., Ribatet, M., Robert, C. P., & Estoup, A. (2018). ABC random forests for Bayesian parameter inference. Bioinformatics, 35(10), 1720-1728. doi: https://dx.doi.org/10.1093/bioinformatics/bty867 [7] Estoup, A., Jarne, P., & Cornuet, J. M. (2002). Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Molecular ecology, 11(9), 1591-1604. doi: https://dx.doi.org/10.1046/j.1365-294X.2002.01576.x [8] Moodley, Y. et al. (2018). Contrasting evolutionary history, anthropogenic declines and genetic contact in the northern and southern white rhinoceros (Ceratotherium simum). Proceedings of the Royal Society B, 285(1890), 20181567. doi: https://dx.doi.org/10.1098/rspb.2018.1567 [9] Kröpelin, S. et al. (2008). Climate-driven ecosystem succession in the Sahara: the past 6000 years. science, 320(5877), 765-768. doi: https://dx.doi.org/10.1126/science.1154913 [10] Maley, J. et al. (2018). Late Holocene forest contraction and fragmentation in central Africa. Quaternary Research, 89(1), 43-59. doi: https://dx.doi.org/10.1017/qua.2017.97 [11] Beheregaray, L. B. (2008). Twenty years of phylogeography: the state of the field and the challenges for the Southern Hemisphere. Molecular Ecology, 17(17), 3754-3774. doi: https://dx.doi.org/10.1111/j.1365-294X.2008.03857.x [12] Dubey, S., & Shine, R. (2012). Are reptile and amphibian species younger in the Northern Hemisphere than in the Southern Hemisphere?. Journal of evolutionary biology, 25(1), 220-226. doi: https://dx.doi.org/10.1111/j.1420-9101.2011.02417.x ***** A video about this preprint is available here: | A young age of subspecific divergence in the desert locust Schistocerca gregaria, inferred by ABC Random Forest | Marie-Pierre Chapuis, Louis Raynal, Christophe Plantamp, Christine N. Meynard, Laurence Blondin, Jean-Michel Marin, Arnaud Estoup | <p>Dating population divergence within species from molecular data and relating such dating to climatic and biogeographic changes is not trivial. Yet it can help formulating evolutionary hypotheses regarding local adaptation and future responses t... | | Bioinformatics & Computational Biology, Evolutionary Applications, Phylogeography & Biogeography, Population Genetics / Genomics | Takeshi Kawakami | 2019-06-20 10:31:15 | ||
11 Dec 2020

Quantifying transmission dynamics of acute hepatitis C virus infections in a heterogeneous population using sequence dataPhylodynamics of hepatitis C virus reveals transmission dynamics within and between risk groups in LyonRecommended by David Rasmussen based on reviews by Chris Wymant and Louis DuPlessisGenomic epidemiology seeks to better understand the transmission dynamics of infectious pathogens using molecular sequence data. Phylodynamic methods have given genomic epidemiology new power to track the transmission dynamics of pathogens by combining phylogenetic analyses with epidemiological modeling. In recent year, applications of phylodynamics to chronic viral infections such as HIV and hepatitis C virus (HVC) have provided some of the best examples of how phylodynamic inference can provide valuable insights into transmission dynamics within and between different subpopulations or risk groups, allowing for more targeted interventions. References [1] Rasmussen, D. A., Volz, E. M., and Koelle, K. (2014). Phylodynamic inference for structured epidemiological models. PLoS Comput Biol, 10(4), e1003570. doi: https://doi.org/10.1371/journal.pcbi.1003570 | Quantifying transmission dynamics of acute hepatitis C virus infections in a heterogeneous population using sequence data | Gonche Danesh, Victor Virlogeux, Christophe Ramière, Caroline Charre, Laurent Cotte, Samuel Alizon | <p>Opioid substitution and syringes exchange programs have drastically reduced hepatitis C virus (HCV) spread in France but HCV sexual transmission in men having sex with men (MSM) has recently arisen as a significant public health concern. The fa... | | Evolutionary Epidemiology, Phylogenetics / Phylogenomics | David Rasmussen | 2019-07-11 13:37:23 | ||
03 Apr 2020

Evolution at two time-frames: ancient and common origin of two structural variants involved in local adaptation of the European plaice (Pleuronectes platessa)Genomic structural variants involved in local adaptation of the European plaiceRecommended by Maren Wellenreuther based on reviews by 3 anonymous reviewersAwareness has been growing that structural variants in the genome of species play a fundamental role in adaptive evolution and diversification [1]. Here, Le Moan and co-authors [2] report empirical genomic-wide SNP data on the European plaice (Pleuronectes platessa) across a major environmental transmission zone, ranging from the North Sea to the Baltic Sea. Regions of high linkage disequilibrium suggest the presence of two structural variants that appear to have evolved 220 kya. These two putative structural variants show weak signatures of isolation by distance when contrasted against the rest of the genome, but the frequency of the different putative structural variants appears to co-vary in some parts of the studied range with the environment, indicating the involvement of both selective and neutral processes. This study adds to the mounting body of evidence that structural genomic variants harbour significant information that allows species to respond and adapt to the local environmental context. References [1] Wellenreuther, M., Mérot, C., Berdan, E., & Bernatchez, L. (2019). Going beyond SNPs: the role of structural genomic variants in adaptive evolution and species diversification. Molecular ecology, 28(6), 1203-1209. doi: 10.1111/mec.15066 | Evolution at two time-frames: ancient and common origin of two structural variants involved in local adaptation of the European plaice (Pleuronectes platessa) | Alan Le Moan, Dorte Bekkevold & Jakob Hemmer-Hansen | <p>Changing environmental conditions can lead to population diversification through differential selection on standing genetic variation. Structural variant (SV) polymorphisms provide examples of ancient alleles that in time become associated with... | | Adaptation, Hybridization / Introgression, Population Genetics / Genomics, Speciation | Maren Wellenreuther | 2019-07-13 12:44:01 | ||
18 Jun 2020
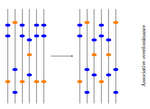
Towards an improved understanding of molecular evolution: the relative roles of selection, drift, and everything in betweenMolecular evolution through the joint lens of genomic and population processes.Recommended by Guillaume Achaz based on reviews by Benoit Nabholz and 1 anonymous reviewerIn their perspective article, F Pouyet and KJ Gilbert (2020), propose an interesting overview of all the processes that sculpt patterns of molecular evolution. This well documented article covers most (if not all) important facets of the recurrent debate that has marked the history of molecular evolution: the relative importance of natural selection and neutral processes (i.e. genetic drift). I particularly enjoyed reading this review, that instead of taking a clear position on the debate, catalogs patiently every pieces of information that can help understand how patterns we observed at the genome level, can be understood from a selectionnist point of view, from a neutralist one, and, to quote their title, from "everything in between". The review covers the classical objects of interest in population genetics (genetic drift, selection, demography and structure) but also describes several genomic processes (meiotic drive, linked selection, gene conversion and mutation processes) that obscure the interpretation of these population processes. The interplay between all these processes is very complex (to say the least) and have resulted in many cases in profound confusions while analyzing data. It is always very hard to fully acknowledge our ignorance and we have many times payed the price of model misspecifications. This review has the grand merit to improve our awareness in many directions. Being able to cover so many aspects of a wide topic, while expressing them simply and clearly, connecting concepts and observations from distant fields, is an amazing "tour de force". I believe this article constitutes an excellent up-to-date introduction to the questions and problems at stake in the field of molecular evolution and will certainly also help established researchers by providing them a stimulating overview supported with many relevant references. References [1] Pouyet F, Gilbert KJ (2020) Towards an improved understanding of molecular evolution: the relative roles of selection, drift, and everything in between. arXiv:1909.11490 [q-bio]. ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. url:https://arxiv.org/abs/1909.11490 | Towards an improved understanding of molecular evolution: the relative roles of selection, drift, and everything in between | Fanny Pouyet and Kimberly J. Gilbert | <p>A major goal of molecular evolutionary biology is to identify loci or regions of the genome under selection versus those evolving in a neutral manner. Correct identification allows accurate inference of the evolutionary process and thus compreh... | | Genome Evolution, Population Genetics / Genomics | Guillaume Achaz | 2019-09-26 10:58:10 |




