
Latest recommendations

| Id | Title▲ | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
26 Aug 2021
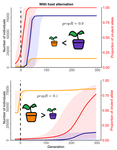
Impact of ploidy and pathogen life cycle on resistance durabilityDurability of plant resistance to diploid pathogenRecommended by Hirohisa Kishino based on reviews by Loup Rimbaud and 1 anonymous reviewerDurability of plant resistance to diploid pathogen Hirohisa Kishino Based on the population genetic and epidemiologic model, Saubin et al. (2021) report that the resistant hosts generated by the breeding based on the gene-for-gene interaction is durable much longer against diploid pathogens than haploid pathogens. The avr allele of pathogen that confers the resistance is genetically recessive. The heterozygotes are not recognized by the resistant hosts and only the avr/avr homozygote is adaptive. As a result, the trajectory of avr allele frequency becomes more stochastic due to genetic drift. Although the paper focuses on the evolution of standing polymorphism, it seems obvious that the adaptive mutations in pathogen have much larger probability of being deleted from the population because the individuals own the avr allele mostly in the form of heterozygote at the initial phase after the mutation. Since only few among many models of plant resistance deployment study the case of diploid pathogen and the contribution of the pathogen life cycle, this work will add an important intellect to the literature (Rimbaud et al. 2021). From the study of host-parasite interaction in flax rust Melampsora lini, Flor (1942, 1955) showed that the host resistance is formed by the interaction of a host resistance gene and a corresponding pathogen gene. This gene-for-gene hypothesis has been supported by experimental evidence and has served as a basis of the methods of molecular breeding targeting the dominant R genes. However, modern agriculture provides the pathogen populations with the homogeneous environments and laid strong selection pressure on them. As a result, the newly developed resistant plants face the risk of immediate resistance breakdown (Möller and Stukenbrock 2017). Currently, quantitative resistance is getting attention as characters as a potential target for long-life (mild) resistant breeds (Lannou, 2012). They are polygenic and controlled partly by the same genes that mediate qualitative resistance but mostly by the genes that encode defense-related outputs such as strengthening of the cell wall or defense compound biosynthesis (Corwin and Kliebenstein, 2017). Progress of molecular genetics may overcome the technical difficulty (Bakkeren and Szabo, 2020). Saubin et al. (2021) notes that the pattern of genetic inheritance of the pathogen counterparts that respond to the host traits is crucial regarding with the durability of the resistant hosts. The resistance traits for which avr alleles are predicted to be recessive may be the targets of breeding. References Bakkeren, G., and Szabo, L. J. (2020) Progress on molecular genetics and manipulation of rust fungi. Phytopathology, 110, 532-543. https://doi.org/10.1094/PHYTO-07-19-0228-IA Corwin, J. A., and Kliebenstein, D. J. (2017) Quantitative resistance: more than just perception of a pathogen. The Plant Cell, 29, 655-665. https://doi.org/10.1105/tpc.16.00915 Flor, H. H. (1942) Inheritance of pathogenicity in a cross between physiological races 22 and 24 of Melampsova lini. Phytopathology, 35. Abstract. Flor, H. H. (1955) Host-parasite interactions in flax rust-its genetics and other implications. Phytopathology, 45, 680-685. Lannou, C. (2012) Variation and selection of quantitative traits in plant pathogens. Annual review of phytopathology, 50, 319-338. https://doi.org/10.1146/annurev-phyto-081211-173031 Möller, M. and Stukenbrock, E. H. (2017) Evolution and genome architecture in fungal plant pathogens. Nature Reviews Microbiology. 15, 756–771. https://doi.org/10.1038/nrmicro.2017.76 Rimbaud, L., Fabre, F., Papaïx, J., Moury, B., Lannou, C., Barrett, L. G., and Thrall, P. H. (2021) Models of Plant Resistance Deployment. Annual Review of Phytopathology, 59. https://doi.org/10.1146/annurev-phyto-020620-122134 Saubin, M., De Mita, S., Zhu, X., Sudret, B. and Halkett, F. (2021) Impact of ploidy and pathogen life cycle on resistance durability. bioRxiv, 2021.05.28.446112, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.05.28.446112 | Impact of ploidy and pathogen life cycle on resistance durability | Méline Saubin, Stephane De Mita, Xujia Zhu, Bruno Sudret, Fabien Halkett | <p>The breeding of resistant hosts based on the gene-for-gene interaction is crucial to address epidemics of plant pathogens in agroecosystems. Resistant host deployment strategies are developed and studied worldwide to decrease the probability of... | | Evolutionary Applications, Evolutionary Epidemiology | Hirohisa Kishino | 2021-06-03 07:58:16 | ||
22 Feb 2023
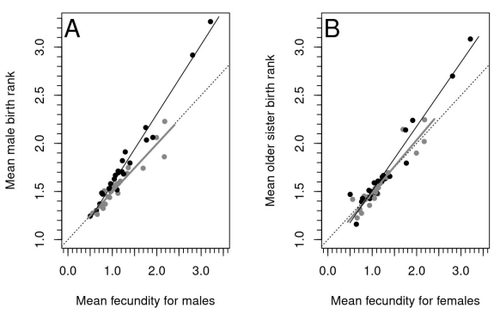
Increased birth rank of homosexual males: disentangling the older brother effect and sexual antagonism hypothesisEvolutionary or proximal explanations for human male homosexual mate preference?Recommended by Jacqui A. Shykoff based on reviews by Ray Blanchard and 1 anonymous reviewer based on reviews by Ray Blanchard and 1 anonymous reviewer
Natural populations do not consist of only perfectly adapted individuals. If they did, of course, there would be no fodder for evolution by natural selection. And natural selection is operating all the time, winnowing out less well adapted phenotypes through differential reproduction and survival. Demonstrations of natural selection modifying characters-state distributions to bring phenotypes closer to their optima abound in the evolution literature, with examples of short- and long-term changes in phenotype and allele frequencies. However, evolutionary biologists know that populations cannot reach their adaptive peaks. Natural selection is tracking a moving target, always with some generations of lag time. The adaptive landscape is multidimensional, so the optimal combination of multiple character states may be impossible because of constraints and trade-offs. Natural selection does not operate alone or in isolation – new mutations and migrants that were selected under other conditions will inject locally non-adaptive genetic variation and genetic drift can change allele frequencies in random directions. We understand these processes that generate and maintain less advantageous variants on a continuous gradient from an optimal phenotype in a fitness landscape. More puzzling are heritable polymorphisms with distinct morphologies, physiologies or behaviours maintained in populations despite their measurably lower reproductive success. But a complete model of evolution must also be able to accommodate these Darwinian paradoxes. Raymond et al. (2023) investigate one such Darwinian paradox: In humans, male homosexual mate preference is heritable and is associated with a large reduction in offspring production but nonetheless occurs at relatively high frequencies in most human populations. Furthermore, multiple studies have found that homosexual men come from families that are, on average, larger than those of heterosexual men and that homosexual men have, on average, higher birth rank than do heterosexual men, i.e., having more older siblings and, particularly, more older brothers. Two types of mechanisms consistent with these observations have been proposed: 1) An evolutionary mechanism of sex-antagonistic pleiotropy, whereby highly fecund mothers are more likely to produce homosexual sons, and 2) A mechanistic explanation whereby successive male pregnancies alter the uterine environment by increasing the probability of an immune reaction by the mother to her male fetus, altering development of sexually dimorphic brain structures relevant to sexual orientation. In this article, the authors explore these two mechanisms of sex-antagonistic effects (AE) and fraternal birth order effects (FBOE) and test how well they account for patterns of male homosexuality in population and family data. Clearly, these two effects are somewhat confounded because high birth ranks can only occur in large families. If, indeed, the probability of male homosexuality increases with increasing numbers of (maternal) older brothers, homosexual males will be more common in larger families. Similarly, if high female fecundity leads to a higher probability of male homosexuality via sex-antagonistic effects, homosexual males will, on average, have more older brothers. To disentangle the actions of these two effects the authors modelled the relationship between birth rank and population fecundity and investigated whether AE or FBOE modified this relationship for homosexual men. Simulation results were compared with aggregated population data from 13 countries. Family data on individuals’ sexual preference, birth rank and number of male and female siblings from France, Greece and Indonesia were analysed with generalised linear models and Bayesian approaches to test for a signal of AE or FBOE. These analyses revealed a significant older-brother effect (FBOE) explaining patterns of occurrence of homosexuality in population and family data but no significant independent sex-antagonistic effect (AE). Thus larger family sizes of homosexual men appear due to the older-brother effect, with individuals of high birth rank coming necessarily from large sibships. The simulation approach also revealed that modelling a fraternal birth order effect (FBOE), such that individuals with more older brothers are more likely to be homosexual, generates an artefactual older sister effect simply because homosexual men are overrepresented at higher birth ranks. Older-sister effects reported in the literature may, therefore, be statistical artefacts of an underlying older-brother effect. This paper is interesting for a number of reasons. It does an excellent job of explaining, identifying and dealing with estimation biases and testing for artefactual relationships generated by collinearity. It applies state-of-the art analytical/statistical tools. It breaks down two colinear effects and shows that only one really explains phenotypic variation. This is a great example of how to disentangle correlated variables that may or may not both contribute to trait variation. But most intriguingly, we are left without evidence for an evolutionary mechanism that compensates the large fitness cost associated with male homosexuality in humans. How can we explain high heritability maintained in the face of strong directional selection that should erode heritable genetic variation? The usual suspects include cryptic compensatory mechanisms yet to be discovered or flawed estimates of selection or heritability. For example, data on heritability of male homosexual mate preference in humans come from twin studies and twins share birth rank as well as alleles. Thus it is possible that heritability is over-estimated, including the environmental component associated with birth rank. If, as the authors demonstrate here, birth rank is the strongest predictor of male homosexual mate preference, selection may be acting on a non-heritable plastic component of phenotypic variation. This could explain why heritable variation is not exhausted by selection, rendering the paradox less paradoxical, but fails to provide an adaptive explanation for the maintenance of male homosexual mate preference. References Raymond M., Turek D., Durand V., Nila S., Suryobroto B., Vadez J., Barthes J., Apostolou M. and Crochet P.-A. (2023) Increased birth rank of homosexual males: disentangling the older brother effect and sexual antagonism hypothesis. bioRxiv, 2022.02.22.481477, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.02.22.481477 | Increased birth rank of homosexual males: disentangling the older brother effect and sexual antagonism hypothesis | Michel Raymond, Daniel Turek, Valerie Durand, Sarah Nila, Bambang Suryobroto, Julien Vadez, Julien Barthes, Menelaos Apostolou, Pierre-André Crochet | <p style="text-align: justify;">Male homosexual orientation remains a Darwinian paradox, as there is no consensus on its evolutionary (ultimate) determinants. One intriguing feature of homosexual men is their higher male birth rank compared to het... | | Life History, Other, Phenotypic Plasticity, Reproduction and Sex | Jacqui A. Shykoff | 2022-03-03 11:28:44 | ||
29 Nov 2023
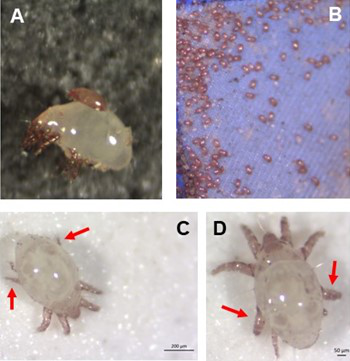
Individual differences in developmental trajectory leave a male polyphenic signature in bulb mite populationsWhat determines whether to scramble or fight in male bulb mitesRecommended by Ulrich Karl Steiner based on reviews by 2 anonymous reviewers
A classic textbook example in evolutionary ecology for phenotypic plasticity—the expression of different phenotypes by a genotype under different environmental conditions—is on Daphnia (Dodson 1989). If various species of this small crustacean are exposed to predation risk or cues thereof, their offspring show induced defense phenotypes including helmets, neck teeth, or head- and tail-spines. These induced morphological changes lower the risk of being eaten by a predator. As in Daphnia, induction can span over generations, while other induced phenotypic plastic changes are almost instantaneous, including many responses in physiology and behaviour (Gabriel et al. 2005). Larvae male bulb mites also show plasticity in morphologies throughout development. They can develop into a costly adult fighter morphology or a less costly, but vulnerable, scrambler type. The question Deere and Smallegange (Deere & Smallegange 2023) address is whether male bulb mite larvae can anticipate which type will likely be adaptive once they become adult, or alternatively, whether the resource availability or population density they experience during their larvae phase determines frequencies of adult scrambler and fighter types. They explore this question through experimental evolution, by removing different fractions of developing intermediate larva types. They thereby manipulate the stage structure of populations and alter selective forces on these stages. The potential shift of fixed genetics, imposed by the experimental selection regimes, is evaluated by fitness assays in green garden experiments. The exciting extension to classical experiments on phenotypic plasticity is that the authors aim at exploring eco-evolutionary feedbacks experimentally in a system that is a little more complex than basic host-parasite or predator-prey systems. The latter involve, for instance, rotifer-algae dynamics (Yoshida et al. 2003; Becks et al. 2012) or similar simple lab systems for which eco-evolutionary feedbacks have been demonstrated. The challenge for the exploration of more complex systems is revealed in the study by Deere and Smallegange. Their findings suggest that the frequencies of adult male morphotype is triggered by the environmental condition (nutrient availability) during the larval phase, i.e. a simple environmental induced plastic response. No fixed genetic shift in determining adult morphotype frequencies occurs. The trigger at the larva phase remains also not perfectly determined in their experiments, as population density and resource (food) availability are partly confounded. Additional complexity and selective aspects come into play in this system, as the targeted developmental stages that develop into fighter male morphs are also dispersal morphs. If selection on dispersal to avoid residing in food-limited environments is strong, triggering genetic shifts in fighter morphs by local population structure might be hard to experimentally achieve. Small sample sizes limit conclusions on complex interactions among duration of the experiment, population size, developmental stage types, and adult fighter frequencies. The presented study (Deere & Smallegange 2023) helps to bridge theoretical predictions and empirical evidence for eco-evolutionary feedbacks that goes beyond simple ecological-driven changes in population dynamics (Govaert et al. 2019).
References
Becks, L., Ellner, S.P., Jones, L.E. & Hairston, N.G. (2012). The functional genomics of an eco-evolutionary feedback loop: linking gene expression, trait evolution, and community dynamics. Ecol. Lett., 15, 492-501. | Individual differences in developmental trajectory leave a male polyphenic signature in bulb mite populations | Jacques A. Deere & Isabel M. Smallegange | <p style="text-align: justify;">Developmental plasticity alters phenotypes and can in that way change the response to selection. When alternative phenotypes show different life history trajectories, developmental plasticity can also affect, and be... | | Evolutionary Ecology, Life History, Phenotypic Plasticity, Sexual Selection | Ulrich Karl Steiner | 2023-02-07 12:14:33 | ||
28 Feb 2018

Insects and incest: sib-mating tolerance in natural populations of a parasitoid waspIncestuous insects in nature despite occasional fitness costsRecommended by Caroline Nieberding and Bertanne Visser based on reviews by 2 anonymous reviewersInbreeding, or mating between relatives, generally lowers fitness [1]. Mating between genetically similar individuals can result in higher levels of homozygosity and consequently a higher frequency with which recessive disease alleles may be expressed within a population. Reduced fitness as a consequence of inbreeding, or inbreeding depression, can vary between individuals, sexes, populations and species [2], but remains a pervasive challenge for many organisms with small local population sizes, including humans [3]. But all is not lost for individuals within small populations, because an array of mechanisms can be employed to evade the negative effects of inbreeding [4], including sib-mating avoidance and dispersal [5, 6]. Despite thorough investigation of inbreeding and sib-mating avoidance in the laboratory, only very few studies have ventured into the field besides studies on vertebrates and eusocial insects. The study of Collet et al. [7] is a surprising exception, where the effect of male density and frequency of relatives on inbreeding avoidance was tested in the laboratory, after which robust field collections and microsatellite genotyping were used to infer relatedness and dispersal in natural populations. The parasitic wasp Venturia canescens is an excellent model system to study inbreeding, because mating success was previously found to decrease with increasing relatedness between mates in the laboratory [8] and this species thus suffers from inbreeding depression [9–11]. The authors used an elegant design combining population genetics and model simulations to estimate relatedness of mating partners in the field and compared that with a theoretical distribution of potential mate encounters when random mating is assumed. One of the most important findings of this study is that mating between siblings is not avoided in this species in the wild, despite negative fitness effects when inbreeding does occur. Similar findings were obtained for another insect species, the field cricket Gryllus campestris [12], which leaves us to wonder whether inbreeding tolerance could be more common in nature than currently appreciated. The authors further looked into sex-specific dispersal patterns between two patches located a few hundred meters apart. Females were indeed shown to be more related within a patch, but no genetic differences were observed between males, suggesting that V. canescens males more readily disperse. Moreover, microsatellite data at 18 different loci did not reveal genetic differentiation between populations approximately 300 kilometers apart. Gene flow is thus occurring over considerable distances, which could play an important role in the ability of this species to avoid negative fitness consequences of inbreeding in nature. Another interesting aspect of this work is that discrepancies were found between laboratory- and field-based data. What is the relevance of laboratory-based experiments if they cannot predict what is happening in the wild? Many, if not most, biologists (including us) bring our model system into the laboratory to control, at least to some extent, the plethora of environmental factors that could potentially affect our system (in ways that we do not want). Most behavioral studies on mating patterns and sexual selection are conducted in standardized laboratory conditions, but sexual selection is in essence social selection, because an individual’s fitness is partly determined by the phenotype of its social partners (i.e. the social environment) [13]. The social environment may actually dictate the expression of female mate choice and it is unclear how potential laboratory-induced social biases affect mating outcome. In V. canescens, findings using field-caught individuals paint a completely opposite picture of what was previously shown in the laboratory, i.e. sib-avoidance is not taking place in the field. It is likely that density, level of relatedness, sex ratio in the field, and/or the size of experimental arenas in the lab are all factors affecting mate selectivity, as we have previously shown in a butterfly [14–16]. If females, for example, typically only encounter a few males in sequence in the wild, it may be problematic for them to express choosiness when confronted simultaneously with two or more males in the laboratory. A recent study showed that, in the wild, female moths take advantage of staying in groups to blur male choosiness [17]. It is becoming more and more clear that what we observe in the laboratory may not actually reflect what is happening in nature [18]. Instead of ignoring the species-specific life history and ecological features of our favorite species when conducting lab experiments, we suggest that it is time to accept that we now have the theoretical foundations to tease apart what in this “environmental noise” actually shapes sexual selection in nature. Explicitly including ecology in studies on sexual selection will allow us to make more meaningful conclusions, i.e. rather than “this is what may happen in the wild”, we would be able to state “this is what often happens in nature”. References [1] Charlesworth D & Willis JH. 2009. The genetics of inbreeding depression. Nat. Rev. Genet. 10: 783–796. doi: 10.1038/nrg2664 | Insects and incest: sib-mating tolerance in natural populations of a parasitoid wasp | Marie Collet, Isabelle Amat, Sandrine Sauzet, Alexandra Auguste, Xavier Fauvergue, Laurence Mouton, Emmanuel Desouhant | <p>This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (http://dx.doi.org/10.24072/pci.evolbiol.100047) 1. Sib-mating avoidance is a pervasive behaviour that likely evolves in species subject to inbreeding dep... | | Behavior & Social Evolution, Evolutionary Ecology, Sexual Selection | Caroline Nieberding | 2017-07-28 09:23:20 | ||
04 Mar 2024
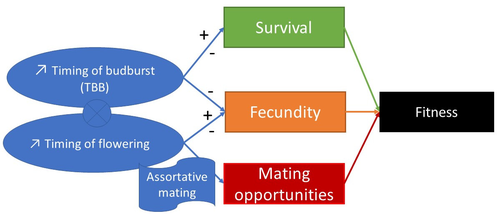
Interplay between fecundity, sexual and growth selection on the spring phenology of European beech (Fagus sylvatica L.).Interplay between fecundity, sexual and growth selection on the spring phenology of European beech (Fagus sylvatica L.)Recommended by Santiago C. Gonzalez-Martinez based on reviews by 2 anonymous reviewers
Starting with the seminar paper by Lande & Arnold (1983), several studies have addressed phenotypic selection in natural populations of a wide variety of organisms, with a recent renewed interest in forest trees (e.g., Oddou-Muratorio et al. 2018; Alexandre et al. 2020; Westergren et al. 2023). Because of their long generation times, long-lived organisms such as forest trees may suffer the most from maladaptation due to climate change, and whether they will be able to adapt to new environmental conditions in just one or a few generations is hotly debated. In this study, Oddou-Muratorio and colleagues (2024) extend the current framework to add two additional selection components that may alter patterns of fecundity selection and the estimation of standard selection gradients, namely sexual selection (evaluated as differences in flowering phenology conducting to assortative mating) and growth (viability) selection. Notably, the study is conducted in two contrasted environments (low vs high altitude populations) providing information on how the environment may modulate selection patterns in spring phenology. Spring phenology is a key adaptive trait that has been shown to be already affected by climate change in forest trees (Alberto et al. 2013). While fecundity selection for early phenology has been extensively reported before (see Munguía-Rosas et al. 2011), the authors found that this kind of selection can be strongly modulated by sexual selection, depending on the environment. Moreover, they found a significant correlation between early phenology and seedling growth in a common garden, highlighting the importance of this trait for early survival in European beech. As a conclusion, this original research puts in evidence the need for more integrative approaches for the study of natural selection in the field, as well as the importance of testing multiple environments and the relevance of common gardens to further evaluate phenotypic changes due to real-time selection. PS: The recommender and the first author of the preprint have shared authorship in a recent paper in a similar topic (Westergren et al. 2023). Nevertheless, the recommender has not contributed in any way or was aware of the content of the current preprint before acting as recommender, and steps have been taken for a fair and unpartial evaluation. References Alberto, F. J., Aitken, S. N., Alía, R., González‐Martínez, S. C., Hänninen, H., Kremer, A., Lefèvre, F., Lenormand, T., Yeaman, S., Whetten, R., & Savolainen, O. (2013). Potential for evolutionary responses to climate change - evidence from tree populations. Global Change Biology, 19(6), 1645‑1661. Oddou-Muratorio S, Bontemps A, Gauzere J, Klein E (2024) Interplay between fecundity, sexual and growth selection on the spring phenology of European beech (Fagus sylvatica L.). bioRxiv, 2023.04.27.538521, ver. 2 peer-reviewed and recommended by Peer Community In Evolutionary Biology https://doi.org/10.1101/2023.04.27.538521 Oddou-Muratorio, S., Gauzere, J., Bontemps, A., Rey, J.-F., & Klein, E. K. (2018). Tree, sex and size: Ecological determinants of male vs. female fecundity in three Fagus sylvatica stands. Molecular Ecology, 27(15), 3131‑3145. | Interplay between fecundity, sexual and growth selection on the spring phenology of European beech (*Fagus sylvatica* L.). | Sylvie Oddou-Muratorio, Aurore Bontemps, Julie Gauzere, Etienne Klein | <p>Background: Plant phenological traits such as the timing of budburst or flowering can evolve on ecological timescales through response to fecundity and viability selection. However, interference with sexual selection may arise from assortative ... | | Adaptation, Evolutionary Ecology, Quantitative Genetics, Reproduction and Sex, Sexual Selection | Santiago C. Gonzalez-Martinez | 2023-05-02 11:57:23 | ||
02 Sep 2022

Introgression between highly divergent sea squirt genomes: an adaptive breakthrough?A match made in the Anthropocene: human-mediated adaptive introgression across a speciation continuumRecommended by Fernando Racimo based on reviews by Michael Westbury, Andrew Foote and Erin CalfeeThe long-distance transport and introduction of new species by humans is increasingly leading divergent lineages to interact, and sometimes interbreed, even after thousands or millions of years of separation. It is thus of prime importance to understand the consequences of these contemporary admixture events on the evolutionary fitness of interacting organisms, and their ecological implications. Ciona robusta and Ciona intestinalis are two species of sea squirts that diverged between 1.5 and 2 million years ago and recently came into contact again. This occurred through human-mediated introduction of C. robusta (native to the Northwest Pacific) into the range of C. intestinalis (the English channeled Northeast Atlantic). In this study, Fraïsse et al. (2022) follow up on earlier work by Le Moan et al. (2021), who had identified a long genomic hotspot of introgression of C. robusta ancestry segments in chromosome 5 of C. intestinalis. The hotspot bears suggestive evidence of positive selection and the authors aimed to investigate this further using fully phased whole-genome sequences. The authors narrow down on the exact boundaries of the introgressed region, and make a compelling case that it has been the likely target of positive selection after introgression, using various complementary approaches based on patterns of population differentiation, haplotype structure and local levels of diversity in the region. Using extensive demographic modeling, they also show that the introgression event was likely recent (approximately 75 years ago), and distinct from other tracts in the C. intestinalis genome that are likely a product of more ancient episodes of interbreeding in the past 30,000 years. Narrowing down on the potential drivers of selection, the authors show that candidate SNPs in the region overlap with the cytochrome family 2 subfamily U gene - involved in the detoxification of exogenous compounds - potentially reflecting adaptation to chemicals encountered in the sea squirt's environment. There also appears to be copy number variation at the candidate SNPs, which provides clues into the adaptation mechanism in the region. All reviewers agreed that the work carried out by the authors is elegant and the results are robustly supported and well presented. In a round of reviews, various clarifications of the manuscript were suggested by the reviewers, including on the quality of the newly generated sequencing data, and some suggestions for qualifications on the conclusions reached by the authors as well as changes in the figures to increase their clarity. The authors addressed the different concerns of the reviewers, and the new version is much improved. This study into human-mediated introgression and its consequences for adaptation is, in my view, both well thought-out and executed. I therefore provide an enthusiastic recommendation of this manuscript. References Fraïsse C, Le Moan A, Roux C, Dubois G, Daguin-Thiébaut C, Gagnaire P-A, Viard F and Bierne N (2022) Introgression between highly divergent sea squirt genomes: an adaptive breakthrough? bioRxiv, 2022.03.22.485319, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.03.22.485319 Le Moan A, Roby C, Fraïsse C, Daguin-Thiébaut C, Bierne N, Viard F (2021) An introgression breakthrough left by an anthropogenic contact between two ascidians. Molecular Ecology, 30, 6718–6732. https://doi.org/10.1111/mec.16189 | Introgression between highly divergent sea squirt genomes: an adaptive breakthrough? | Christelle Fraïsse, Alan Le Moan, Camille Roux, Guillaume Dubois, Claire Daguin-Thiébaut, Pierre-Alexandre Gagnaire, Frédérique Viard, Nicolas Bierne | <p style="text-align: justify;">Human-mediated introductions are reshuffling species distribution on a global scale. Consequently, an increasing number of allopatric taxa are now brought into contact, promoting introgressive hybridization between ... | | Adaptation, Hybridization / Introgression, Population Genetics / Genomics | Fernando Racimo | 2022-04-14 15:30:42 | ||
28 Aug 2019

Is adaptation limited by mutation? A timescale-dependent effect of genetic diversity on the adaptive substitution rate in animalsTo tinker, evolution needs a supply of spare partsRecommended by Georgii Bazykin based on reviews by Konstantin Popadin, David Enard and 1 anonymous reviewerIs evolution adaptive? Not if there is no variation for natural selection to work with. Theory predicts that how fast a population can adapt to a new environment can be limited by the supply of new mutations coming into it. This supply, in turn, depends on two things: how often mutations occur and in how many individuals. If there are few mutations, or few individuals in whom they can originate, individuals will be mostly identical in their DNA, and natural selection will be impotent. References [1] G, J. A., Visser, M. de, Zeyl, C. W., Gerrish, P. J., Blanchard, J. L., and Lenski, R. E. (1999). Diminishing Returns from Mutation Supply Rate in Asexual Populations. Science, 283(5400), 404–406. doi: 10.1126/science.283.5400.404 | Is adaptation limited by mutation? A timescale-dependent effect of genetic diversity on the adaptive substitution rate in animals | Marjolaine Rousselle, Paul Simion, Marie-Ka Tilak, Emeric Figuet, Benoit Nabholz, Nicolas Galtier | <p>Whether adaptation is limited by the beneficial mutation supply is a long-standing question of evolutionary genetics, which is more generally related to the determination of the adaptive substitution rate and its relationship with the effective... | | Adaptation, Evolutionary Theory, Genome Evolution, Molecular Evolution, Population Genetics / Genomics | Georgii Bazykin | 2019-05-21 09:49:16 | ||
29 Nov 2022

Joint inference of adaptive and demographic history from temporal population genomic dataInference of genome-wide processes using temporal population genomic dataRecommended by Aurelien Tellier based on reviews by Lawrence Uricchio and 2 anonymous reviewersEvolutionary genomics, and population genetics in particular, aim to decipher the respective influence of neutral and selective forces shaping genetic polymorphism in a species/population. This is a much-needed requirement before scanning genome data for footprints of species adaptation to their biotic and abiotic environment (Johri et al. 2022). In general, we would like to quantify the proportion of the genome evolving neutrally and under selective (positive, balancing and negative) pressures (Kern and Hahn 2018, Johri et al. 2021). We thus need to understand patterns of linked selection along the genome, that is how the distribution of genetic polymorphisms is shaped by selected sites and the recombination landscape. The present contribution by Pavinato et al. (2022) provides an additional method in the population genomics toolbox to quantify the extent of linked positive and negative selection using temporal data. The availability of genomics data for model and non-model species has led to improvement of the modeling framework for demography and selection (Johri et al. 2022), but also new inference methods making use of the full genome data based on the Sequential Markovian Coalescent (SMC, Li and Durbin 2011), Approximate Bayesian Computation (ABC, Jay et al. 2019), ABC and machine learning (Pudlo et al. 2016, Raynal et al. 2019) or Deep Learning (Sanchez et al. 2021). These methods are based on one sample in time and the use of the coalescent theory to reconstruct the past (demographic) history. However, it is also possible to obtain for many species temporal data sampled over several time points. For species with short generation time (in experimental evolution or monitored populations), one can sample a population every couple of generations as exemplified with Drosophila melanogaster (Bergland et al. 2010). For species with longer generation times that cannot be easily regularly sampled in time, it becomes possible to sequence available specimens from museums (e.g. Cridland et al. 2018) or ancient DNA samples. Methods using temporal data are based on the classical population genomics assumption that demography (migration, population subdivision, population size changes) leaves a genome-wide signal, while selection leaves a localized signal in the close vicinity of the causal mutation. Several methods do assess the demography of a population (change in effective population size, Ne, in time) using temporal data (e.g. Jorde and Ryman 2007) which can be used to calibrate the detection of loci under strong positive selection (Foll et al. 2014). Recently Buffalo and Coop (2020) used genome-wide covariance between allele frequency changes across time samples (and across replicates) to quantify the effects of linked selection over short timescales. In the present contribution, Pavinato et al. (2022) make use of temporal data to draw the joint estimation of demographic and selective parameters using a simulation-based method (ABC-Random Forests). This study by Pavinato et al. (2022) builds a framework allowing to infer the census size of the population in time (N) separately from the effect of genetic drift, which is determined by change in effective population size (Ne) in time, as well estimates of genome-wide parameters of selection. In a nutshell, the authors use a forward simulator and summarize genome data by genomic windows using classic statistics (nucleotide diversity, Tajima’s D, FST, heterozygosity) between time samples and for each sample. They specifically use the distributions (higher moments) of these statistics among all windows. The authors combine as input for the ABC-RF, vectors of summary statistics, model parameters and five latent variables: Ne, the ratio Ne/N, the number of beneficial mutations under strong selection, the average selection coefficient of strongly selected mutations, and the average substitution load. Indeed, the authors are interested in three different types of selection components: 1) the adaptive potential of a population which is estimated as the population mutation rate of beneficial mutations (θb), 2) the number of mutations under strong selection (irrespective of whether they reached fixation or not), and 3) the overall population fitness which is a function of the genetic load. In other words, the novelty of this method is not to focus on the detection of loci under selection, but to infer key parameters/distributions summarizing the genome-wide signal of demography and (positive and negative) selection. As a proof of principle, the authors then apply their method to a dataset of feral populations of honey bees (Apis mellifera) collected in California across many years and recovered from Museum samples (Cridland et al. 2018). The approach yields estimates of Ne which are on the same order of magnitude of previous estimates in hymenopterans, and the authors discuss why the different populations show various values of Ne and N which can be explained by different history of admixture with wild but also domesticated lineages of bees. This study focuses on quantifying the genome-wide joint footprints of demography, and strong positive and negative selection to determine which proportion of the genome evolves neutrally or not. Further application of this method can be anticipated, for example, to study species with ecological and life-history traits which generate discrepancies between census size and Ne, for example for plants with selfing or seed banking (Sellinger et al. 2020), and for which the genome-wide effect of linked selection is not fully understood. References Johri P, Aquadro CF, Beaumont M, Charlesworth B, Excoffier L, Eyre-Walker A, Keightley PD, Lynch M, McVean G, Payseur BA, Pfeifer SP, Stephan W, Jensen JD (2022) Recommendations for improving statistical inference in population genomics. PLOS Biology, 20, e3001669. https://doi.org/10.1371/journal.pbio.3001669 Kern AD, Hahn MW (2018) The Neutral Theory in Light of Natural Selection. Molecular Biology and Evolution, 35, 1366–1371. https://doi.org/10.1093/molbev/msy092 Johri P, Riall K, Becher H, Excoffier L, Charlesworth B, Jensen JD (2021) The Impact of Purifying and Background Selection on the Inference of Population History: Problems and Prospects. Molecular Biology and Evolution, 38, 2986–3003. https://doi.org/10.1093/molbev/msab050 Pavinato VAC, Mita SD, Marin J-M, Navascués M de (2022) Joint inference of adaptive and demographic history from temporal population genomic data. bioRxiv, 2021.03.12.435133, ver. 6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.03.12.435133 Li H, Durbin R (2011) Inference of human population history from individual whole-genome sequences. Nature, 475, 493–496. https://doi.org/10.1038/nature10231 Jay F, Boitard S, Austerlitz F (2019) An ABC Method for Whole-Genome Sequence Data: Inferring Paleolithic and Neolithic Human Expansions. Molecular Biology and Evolution, 36, 1565–1579. https://doi.org/10.1093/molbev/msz038 Pudlo P, Marin J-M, Estoup A, Cornuet J-M, Gautier M, Robert CP (2016) Reliable ABC model choice via random forests. Bioinformatics, 32, 859–866. https://doi.org/10.1093/bioinformatics/btv684 Raynal L, Marin J-M, Pudlo P, Ribatet M, Robert CP, Estoup A (2019) ABC random forests for Bayesian parameter inference. Bioinformatics, 35, 1720–1728. https://doi.org/10.1093/bioinformatics/bty867 Sanchez T, Cury J, Charpiat G, Jay F (2021) Deep learning for population size history inference: Design, comparison and combination with approximate Bayesian computation. Molecular Ecology Resources, 21, 2645–2660. https://doi.org/10.1111/1755-0998.13224 Bergland AO, Behrman EL, O’Brien KR, Schmidt PS, Petrov DA (2014) Genomic Evidence of Rapid and Stable Adaptive Oscillations over Seasonal Time Scales in Drosophila. PLOS Genetics, 10, e1004775. https://doi.org/10.1371/journal.pgen.1004775 Cridland JM, Ramirez SR, Dean CA, Sciligo A, Tsutsui ND (2018) Genome Sequencing of Museum Specimens Reveals Rapid Changes in the Genetic Composition of Honey Bees in California. Genome Biology and Evolution, 10, 458–472. https://doi.org/10.1093/gbe/evy007 Jorde PE, Ryman N (2007) Unbiased Estimator for Genetic Drift and Effective Population Size. Genetics, 177, 927–935. https://doi.org/10.1534/genetics.107.075481 Foll M, Shim H, Jensen JD (2015) WFABC: a Wright–Fisher ABC-based approach for inferring effective population sizes and selection coefficients from time-sampled data. Molecular Ecology Resources, 15, 87–98. https://doi.org/10.1111/1755-0998.12280 Buffalo V, Coop G (2020) Estimating the genome-wide contribution of selection to temporal allele frequency change. Proceedings of the National Academy of Sciences, 117, 20672–20680. https://doi.org/10.1073/pnas.1919039117 Sellinger TPP, Awad DA, Moest M, Tellier A (2020) Inference of past demography, dormancy and self-fertilization rates from whole genome sequence data. PLOS Genetics, 16, e1008698. https://doi.org/10.1371/journal.pgen.1008698 | Joint inference of adaptive and demographic history from temporal population genomic data | Vitor A. C. Pavinato, Stéphane De Mita, Jean-Michel Marin, Miguel de Navascués | <p style="text-align: justify;">Disentangling the effects of selection and drift is a long-standing problem in population genetics. Simulations show that pervasive selection may bias the inference of demography. Ideally, models for the inference o... | | Adaptation, Population Genetics / Genomics | Aurelien Tellier | 2021-10-20 09:41:26 | ||
26 Sep 2017

Lacking conservation genomics in the giant Galápagos tortoiseA genomic perspective is needed for the re-evaluation of species boundaries, evolutionary trajectories and conservation strategies for the Galápagos giant tortoisesRecommended by Michael C. Fontaine based on reviews by 4 anonymous reviewersGenome-wide data obtained from even a small number of individuals can provide unprecedented levels of detail about the evolutionary history of populations and species [1], determinants of genetic diversity [2], species boundaries and the process of speciation itself [3]. Loire and Galtier [4] present a clear example, using the emblematic Galápagos giant tortoise (Chelonoidis nigra), of how multi-species comparative population genomic approaches can provide valuable insights about population structure and species delimitation even when sample sizes are limited but the number of loci is large and distributed across the genome. Galápagos giant tortoises are endemic to the Galápagos Islands and are currently recognized as an endangered, multi-species complex including both extant and extinct taxa. Taxonomic definitions are based on morphology, geographic isolation and population genetic evidence based on short DNA sequences of the mitochondrial genome (mtDNA) and/or a dozen or so nuclear microsatellite loci [5-8]. The species complex enjoys maximal protection. Population recoveries have been quite successful and spectacular conservation programs based on mitochondrial genes and microsatellites are ongoing. This includes for example individual translocations, breeding program, “hybrid” sterilization or removal, and resurrection of extinct lineages). In 2013, Loire et al. [9] provided the first population genomic analyses based on genome scale data (~1000 coding loci derived from blood-transcriptomes) from five individuals, encompassing three putative “species”: Chelonnoidis becki, C. porteri and C. vandenburghi. Their results raised doubts about the validity/accuracy of the currently accepted designations of “genetic distinctiveness”. However, the implications for conservation and management have remained unnoticed. In 2017, Loire and Galtier [4] have re-appraised this issue using an original multi-species comparative population genomic analysis of their previous data set [9]. Based on a comparison of 53 animal species, they show that both the level of genome-wide neutral diversity (πS) and level of population structure estimated using the inbreeding coefficient (F) are much lower than would be expected from a sample covering multiple species. The observed values are more comparable to those typically reported at the “among population” level within a single species such as human (Homo sapiens). The authors go to great length to assess the sensitivity of their method to detect population structure (or lack thereof) and show that their results are robust to potential issues, such as contamination and sequencing errors that can occur with Next Generation Sequencing techniques; and biases related to the small sample size and sub-sampling of individuals. They conclude that published mtDNA and microsatellite-based assessment of population structure and species designations may be biased towards over-splitting. This manuscript is a very good read as it shows the potential of the now relatively affordable genome-wide data for helping to both resolve and clarify population and species boundaries, illuminate demographic trends, evolutionary trajectories of isolated groups, patterns of connectivity among them, and test for evidence of local adaptation and even reproductive isolation. The comprehensive information provided by genome-wide data can critically inform and assist the development of the best strategies to preserve endangered populations and species. Loire and Galtier [4] make a strong case for applying genomic data to the Galápagos giant tortoises, which is likely to redirect conservation efforts more effectively and at lower cost. The case of the Galápagos giant tortoises is certainly a very emblematic example, which will find an echo in many other endangered species conservation programs. References [1] Li H and Durbin R. 2011. Inference of human population history from individual whole-genome sequences. Nature, 475: 493–496. doi: 10.1038/nature10231 [2] Romiguier J, Gayral P, Ballenghien M, Bernard A, Cahais V, Chenuil A, Chiari Y, Dernat R, Duret L, Faivre N, Loire E, Lourenco JM, Nabholz B, Roux C, Tsagkogeorga G, Weber AA-T, Weinert LA, Belkhir K, Bierne N, Glémin S and Galtier N. 2014. Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature, 515: 261–263. doi: 10.1038/nature13685 [3] Roux C, Fraïsse C, Romiguier J, Anciaux Y, Galtier N and Bierne N. 2016. Shedding light on the grey zone of speciation along a continuum of genomic divergence. PLoS Biology, 14: e2000234. doi: 10.1371/journal.pbio.2000234 [4] Loire E and Galtier N. 2017. Lacking conservation genomics in the giant Galápagos tortoise. bioRxiv 101980, ver. 4 of September 26, 2017. doi: 10.1101/101980 [5] Beheregaray LB, Ciofi C, Caccone A, Gibbs JP and Powell JR. 2003. Genetic divergence, phylogeography and conservation units of giant tortoises from Santa Cruz and Pinzón, Galápagos Islands. Conservation Genetics, 4: 31–46. doi: 10.1023/A:1021864214375 [6] Ciofi C, Milinkovitch MC, Gibbs JP, Caccone A and Powell JR. 2002. Microsatellite analysis of genetic divergence among populations of giant Galápagos tortoises. Molecular Ecology, 11: 2265–2283. doi: 10.1046/j.1365-294X.2002.01617.x [7] Garrick RC, Kajdacsi B, Russello MA, Benavides E, Hyseni C, Gibbs JP, Tapia W and Caccone A. 2015. Naturally rare versus newly rare: demographic inferences on two timescales inform conservation of Galápagos giant tortoises. Ecology and Evolution, 5: 676–694. doi: 10.1002/ece3.1388 [8] Poulakakis N, Edwards DL, Chiari Y, Garrick RC, Russello MA, Benavides E, Watkins-Colwell GJ, Glaberman S, Tapia W, Gibbs JP, Cayot LJ and Caccone A. 2015. Description of a new Galápagos giant tortoise species (Chelonoidis; Testudines: Testudinidae) from Cerro Fatal on Santa Cruz island. PLoS ONE, 10: e0138779. doi: 10.1371/journal.pone.0138779 [9] Loire E, Chiari Y, Bernard A, Cahais V, Romiguier J, Nabholz B, Lourenço JM and Galtier N. 2013. Population genomics of the endangered giant Galápagos tortoise. Genome Biology, 14: R136. doi: 10.1186/gb-2013-14-12-r136 | Lacking conservation genomics in the giant Galápagos tortoise | Etienne Loire, Nicolas Galtier | <p>Conservation policy in the giant Galápagos tortoise, an iconic endangered animal, has been assisted by genetic markers for ~15 years: a dozen loci have been used to delineate thirteen (sub)species, between which hybridization is prevented. Here... | | Evolutionary Applications, Population Genetics / Genomics, Speciation, Systematics / Taxonomy | Michael C. Fontaine | 2017-01-21 15:34:00 | ||
11 Oct 2021

Landscape connectivity alters the evolution of density-dependent dispersal during pushed range expansionsPhenotypic evolution during range expansions is contingent on connectivity and density dependenceRecommended by Inês Fragata based on reviews by 3 anonymous reviewersUnderstanding the mechanisms underlying range expansions is key for predicting species distributions in response to environmental changes (such as global warming) and managing the global expansion of invasive species (Parmesan 2006; Suarez & Tsutsui 2008). Traditionally, two types of ecological processes were studied as essential in shaping range expansion: dispersal and population growth. However, ecology and evolution are intertwined in range expansions, as phenotypic evolution of traits involved in demographic and dispersal patterns and processes can affect and be affected by ecological dynamics, representing a full eco-evolutionary loop (Williams et al. 2019; Miller et al. 2020). Range expansions can be characterized by the type of population growth and dispersal, divided into pushed or pulled range expansions. Species that have high dispersal and high population growth at low densities present pulled range expansions (pulled by individuals from the edge populations). In contrast, populations presenting increased growth rate at intermediate densities (due to Allee effects - Allee & Bowen 1932; i.e. where growth rate decreases at lower densities) and high dispersal at high densities present pushed range expansions (driven by individuals from core and intermediate populations) (Gandhi et al. 2016). Importantly, the type of expansion is expected to have very different consequences on the genetic (and therefore) phenotypic composition of core and edge populations. Specifically, genetic variability is expected to be lower in populations experiencing pulled expansions and higher in populations involved in pushed expansions (Gandhi et al. 2016; Miller et al. 2020). However, it is not always possible to distinguish between pulled and pushed expansions, as variation in speed and shape can overlap between the two types. In addition, it is difficult to experimentally manipulate the strength of the Allee effect to create pushed versus pulled expansions. Thus, several critical predictions regarding the genetic and phenotypic composition of pulled and pushed expansions are lacking empirical tests (but see Gandhi et al. 2016). In a previous study, Dahirel et al. (2021a) combined simulations and experimental evolution of the small wasps Trichogramma brassicae to show that low connectivity led to more pushed expansions, and higher connectivity generated more pulled expansions. In accordance with theoretical predictions, this led to reduced genetic diversity in pulled expansions, and the reverse pattern in pushed expansions. However, the question of how pulled and pushed expansions affect trait evolution remained unanswered. In this follow-up study, Dahirel et al. (2021b) tackled this issue and linked the changes in connectivity and type of expansion with the phenotypic evolution of several traits using individuals from their previous experiment. Namely, the authors compared core and edge populations with founder strains to test how evolution in pushed vs. pulled expansions affected wasp size, short movement, fecundity, dispersal, and density dependent dispersal. When density dependence was not accounted for, phenotypic changes in edge populations did not match the expectations from changes in expansion dynamics. This could be due to genetic trade-offs between traits that limit phenotypic evolution (Urquhart & Williams 2021). However, when accounting for density dependent dispersal, Dahirel et al. (2021b) observed that more connected landscapes (with pulled expansions) showed positive density dispersal in core populations and negative density dispersal in edge populations, similarly to other studies (e.g. Fronhofer et al. 2017). Interestingly, in pushed (with lower connectivity) landscapes, such shift was not observed. Instead, edge populations maintained positive density dispersal even after 14 generations of expansion, whereas core populations showed higher dispersal at lower density. The authors suggest that this seemingly contradictory result is due to a combination of three processes: 1) the expansion reduced positive density dispersal in edge populations; 2) reduced connectivity directly increased dispersal costs, increasing high density dispersal; and 3) reduced connectivity indirectly caused demographic stochasticity (and reduced temporal variability in patches) leading to higher dispersal at low density in core populations. However, these results must be taken with a grain of salt, since only one of the four experimental replicates were used in the density dependent dispersal experiment. In range expansions experiments, replication is fundamental, since stochastic processes (such as gene surfing, where alleles maybe rise in frequency due by chance) are prevalent (Miller et al. 2020), and results are highly dependent on sample size, or number of replicate populations analysed. Having said that, results from Dahirel et al. (2021b) highlight the importance to contextualize the management of invasions and species distribution, since it is thought that pulled expansions are more prevalent in nature, but pushed expansions can be more important in scenarios where patchiness is high, such as urban landscapes. Moreover, Dahirel's et al. (2021b) study is a first step showing that accounting for trait density dependence is crucial when following phenotypic evolution during range expansion, and that evolution of density dependent traits may be constrained by landscape conditions. This highlights the need to account for both connectivity and density dependence to draw more accurate predictions on the evolutionary and ecological outcomes of range expansions. Allee WC, Bowen ES (1932) Studies in animal aggregations: Mass protection against colloidal silver among goldfishes. Journal of Experimental Zoology, 61, 185–207. https://doi.org/10.1002/jez.1400610202 Dahirel M, Bertin A, Calcagno V, Duraj C, Fellous S, Groussier G, Lombaert E, Mailleret L, Marchand A, Vercken E (2021a) Landscape connectivity alters the evolution of density-dependent dispersal during pushed range expansions. bioRxiv, 2021.03.03.433752, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.03.03.433752 Dahirel M, Bertin A, Haond M, Blin A, Lombaert E, Calcagno V, Fellous S, Mailleret L, Malausa T, Vercken E (2021b) Shifts from pulled to pushed range expansions caused by reduction of landscape connectivity. Oikos, 130, 708–724. https://doi.org/10.1111/oik.08278 Fronhofer EA, Gut S, Altermatt F (2017) Evolution of density-dependent movement during experimental range expansions. Journal of Evolutionary Biology, 30, 2165–2176. https://doi.org/10.1111/jeb.13182 Gandhi SR, Yurtsev EA, Korolev KS, Gore J (2016) Range expansions transition from pulled to pushed waves as growth becomes more cooperative in an experimental microbial population. Proceedings of the National Academy of Sciences, 113, 6922–6927. https://doi.org/10.1073/pnas.1521056113 Miller TEX, Angert AL, Brown CD, Lee-Yaw JA, Lewis M, Lutscher F, Marculis NG, Melbourne BA, Shaw AK, Szűcs M, Tabares O, Usui T, Weiss-Lehman C, Williams JL (2020) Eco-evolutionary dynamics of range expansion. Ecology, 101, e03139. https://doi.org/10.1002/ecy.3139 Parmesan C (2006) Ecological and Evolutionary Responses to Recent Climate Change. Annual Review of Ecology, Evolution, and Systematics, 37, 637–669. https://doi.org/10.1146/annurev.ecolsys.37.091305.110100 Suarez AV, Tsutsui ND (2008) The evolutionary consequences of biological invasions. Molecular Ecology, 17, 351–360. https://doi.org/10.1111/j.1365-294X.2007.03456.x Urquhart CA, Williams JL (2021) Trait correlations and landscape fragmentation jointly alter expansion speed via evolution at the leading edge in simulated range expansions. Theoretical Ecology. https://doi.org/10.1007/s12080-021-00503-z Williams JL, Hufbauer RA, Miller TEX (2019) How Evolution Modifies the Variability of Range Expansion. Trends in Ecology & Evolution, 34, 903–913. https://doi.org/10.1016/j.tree.2019.05.012 | Landscape connectivity alters the evolution of density-dependent dispersal during pushed range expansions | Maxime Dahirel, Aline Bertin, Vincent Calcagno, Camille Duraj, Simon Fellous, Géraldine Groussier, Eric Lombaert, Ludovic Mailleret, Anaël Marchand, Elodie Vercken | <p style="text-align: justify;">As human influence reshapes communities worldwide, many species expand or shift their ranges as a result, with extensive consequences across levels of biological organization. Range expansions can be ranked on a con... | | Evolutionary Ecology, Experimental Evolution | Inês Fragata | 2021-03-05 17:15:46 |




