
Latest recommendations

| Id | Title | Authors | Abstract▲ | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
14 Apr 2021

Parasitic success and venom composition evolve upon specialization of parasitoid wasps to different host speciesWhat makes a parasite successful? Parasitoid wasp venoms evolve rapidly in a host-specific mannerRecommended by Élio Sucena based on reviews by Simon Fellous, alexandre leitão and 1 anonymous reviewerParasitoid wasps have developed different mechanisms to increase their parasitic success, usually at the expense of host survival (Fellowes and Godfray, 2000). Eggs of these insects are deposited inside the juvenile stages of their hosts, which in turn deploy several immune response strategies to eliminate or disable them (Yang et al., 2020). Drosophila melanogaster protects itself against parasitoid attacks through the production of specific elongated haemocytes called lamellocytes which form a capsule around the invading parasite (Lavine and Strand, 2002; Rizki and Rizki, 1992) and the subsequent activation of the phenol-oxidase cascade leading to the release of toxic radicals (Nappi et al., 1995). On the parasitoid side, robust responses have evolved to evade host immune defenses as for example the Drosophila-specific endoparasite Leptopilina boulardi, which releases venom during oviposition that modifies host behaviour (Varaldi et al., 2006) and inhibits encapsulation (Gueguen et al., 2011; Martinez et al., 2012).
References Cavigliasso, F., Mathé-Hubert, H., Gatti, J.-L., Colinet, D. and Poirié, M. (2021) Parasitic success and venom composition evolve upon specialization of parasitoid wasps to different host species. bioRxiv, 2020.10.24.353417, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.10.24.353417 Cavigliasso, F., Mathé-Hubert, H., Kremmer, L., Rebuf, C., Gatti, J.-L., Malausa, T., Colinet, D., Poiré, M. and Léne. (2019). Rapid and Differential Evolution of the Venom Composition of a Parasitoid Wasp Depending on the Host Strain. Toxins, 11(629). https://doi.org/10.3390/toxins11110629 Colinet, D., Deleury, E., Anselme, C., Cazes, D., Poulain, J., Azema-Dossat, C., Belghazi, M., Gatti, J. L. and Poirié, M. (2013). Extensive inter- and intraspecific venom variation in closely related parasites targeting the same host: The case of Leptopilina parasitoids of Drosophila. Insect Biochemistry and Molecular Biology, 43(7), 601–611. https://doi.org/10.1016/j.ibmb.2013.03.010 Colinet, D., Dubuffet, A., Cazes, D., Moreau, S., Drezen, J. M. and Poirié, M. (2009). A serpin from the parasitoid wasp Leptopilina boulardi targets the Drosophila phenoloxidase cascade. Developmental and Comparative Immunology, 33(5), 681–689. https://doi.org/10.1016/j.dci.2008.11.013 Fellowes, M. D. E. and Godfray, H. C. J. (2000). The evolutionary ecology of resistance to parasitoids by Drosophila. Heredity, 84(1), 1–8. https://doi.org/10.1046/j.1365-2540.2000.00685.x Gueguen, G., Rajwani, R., Paddibhatla, I., Morales, J. and Govind, S. (2011). VLPs of Leptopilina boulardi share biogenesis and overall stellate morphology with VLPs of the heterotoma clade. Virus Research, 160(1–2), 159–165. https://doi.org/10.1016/j.virusres.2011.06.005 Lavine, M. D. and Strand, M. R. (2002). Insect hemocytes and their role in immunity. Insect Biochemistry and Molecular Biology, 32(10), 1295–1309. https://doi.org/10.1016/S0965-1748(02)00092-9 Martinez, J., Duplouy, A., Woolfit, M., Vavre, F., O’Neill, S. L. and Varaldi, J. (2012). Influence of the virus LbFV and of Wolbachia in a host-parasitoid interaction. PloS One, 7(4), e35081. https://doi.org/10.1371/journal.pone.0035081 Nappi, A. J., Vass, E., Frey, F. and Carton, Y. (1995). Superoxide anion generation in Drosophila during melanotic encapsulation of parasites. European Journal of Cell Biology, 68(4), 450–456. Poirié, M., Colinet, D. and Gatti, J. L. (2014). Insights into function and evolution of parasitoid wasp venoms. Current Opinion in Insect Science, 6, 52–60. https://doi.org/10.1016/j.cois.2014.10.004 Rizki, T. M. and Rizki, R. M. (1992). Lamellocyte differentiation in Drosophila larvae parasitized by Leptopilina. Developmental and Comparative Immunology, 16(2–3), 103–110. https://doi.org/10.1016/0145-305X(92)90011-Z Schlenke, T. A., Morales, J., Govind, S. and Clark, A. G. (2007). Contrasting infection strategies in generalist and specialist wasp parasitoids of Drosophila melanogaster. PLoS Pathogens, 3(10), 1486–1501. https://doi.org/10.1371/journal.ppat.0030158 Varaldi, J., Petit, S., Boulétreau, M. and Fleury, F. (2006). The virus infecting the parasitoid Leptopilina boulardi exerts a specific action on superparasitism behaviour. Parasitology, 132(Pt 6), 747–756. https://doi.org/10.1017/S0031182006009930 Yang, L., Qiu, L., Fang, Q., Stanley, D. W. and Gong‐Yin, Y. (2020). Cellular and humoral immune interactions between Drosophila and its parasitoids. Insect Science. https://doi.org/10.1111/1744-7917.12863
| Parasitic success and venom composition evolve upon specialization of parasitoid wasps to different host species | Fanny Cavigliasso, Hugo Mathé-Hubert, Jean-Luc Gatti, Dominique Colinet, Marylène Poirié | <p>Female endoparasitoid wasps usually inject venom into hosts to suppress their immune response and ensure offspring development. However, the parasitoid’s ability to evolve towards increased success on a given host simultaneously with the evolut... | | Experimental Evolution, Species interactions | Élio Sucena | 2020-10-26 15:00:55 | ||
19 Mar 2018

Natural selection on plasticity of thermal traits in a highly seasonal environmentIs thermal plasticity itself shaped by natural selection? An assessment with desert frogsRecommended by Wolf Blanckenhorn based on reviews by Dries Bonte, Wolf Blanckenhorn and Nadia Aubin-HorthIt is well known that climatic factors – most notably temperature, season length, insolation and humidity – shape the thermal niche of organisms on earth through the action of natural selection. But how is this achieved precisely? Much of thermal tolerance is actually mediated by phenotypic plasticity (as opposed to genetic adaptation). A prominent expectation is that environments with greater (daily and/or annual) thermal variability select for greater plasticity, i.e. better acclimation capacity. Thus, plasticity might be selected per se. A Chilean group around Leonardo Bacigalupe assessed natural selection in the wild in one marginal (and extreme) population of the four-eyed frog Pleurodema thaul (Anura: Leptodactylidae) in an isolated oasis in the Atacama Desert, permitting estimation of mortality without much potential of confounding it with migration [1]. Several thermal traits were considered: CTmax – the critical maximal temperature; CTmin – the critical minimum temperature; Tpref – preferred temperature; Q10 – thermal sensitivity of metabolism; and body mass. Animals were captured in the wild and subsequently assessed for thermal traits in the laboratory at two acclimation temperatures (10° & 20°C), defining the plasticity in all traits as the difference between the traits at the two acclimation temperatures. Thereafter the animals were released again in their natural habitat and their survival was monitored over the subsequent 1.5 years, covering two breeding seasons, to estimate viability selection in the wild. The authors found and conclude that, aside from larger body size increasing survival (an unsurprising result), plasticity does not seem to be systematically selected directly, while some of the individual traits show weak signs of selection. Despite limited sample size (ca. 80 frogs) investigated in only one marginal but very seasonal population, this study is interesting because selection on plasticity in physiological thermal traits, as opposed to selection on the thermal traits themselves, is rarely investigated. The study thus also addressed the old but important question of whether plasticity (i.e. CTmax-CTmin) is a trait by itself or an epiphenomenon defined by the actual traits (CTmax and CTmin) [2-5]. Given negative results, the main question could not be ultimately solved here, so more similar studies should be performed. References [1] Bacigalupe LD, Gaitan-Espitia, JD, Barria AM, Gonzalez-Mendez A, Ruiz-Aravena M, Trinder M & Sinervo B. 2018. Natural selection on plasticity of thermal traits in a highly seasonal environment. bioRxiv 191825, ver. 5 peer-reviewed by Peer Community In Evolutionary Biology. doi: 10.1101/191825 | Natural selection on plasticity of thermal traits in a highly seasonal environment | Leonardo Bacigalupe, Juan Diego Gaitan-Espitia, Aura M Barria, Avia Gonzalez-Mendez, Manuel Ruiz-Aravena, Mark Trinder, Barry Sinervo | <p>For ectothermic species with broad geographical distributions, latitudinal/altitudinal variation in environmental temperatures (averages and extremes) are expected to shape the evolution of physiological tolerances and the acclimation capacity ... | | Adaptation, Evolutionary Ecology, Phenotypic Plasticity | Wolf Blanckenhorn | 2017-09-22 23:17:40 | ||
18 Aug 2020
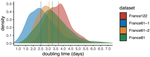
Early phylodynamics analysis of the COVID-19 epidemics in FranceSARS-Cov-2 genome sequence analysis suggests rapid spread followed by epidemic slowdown in FranceRecommended by B. Jesse Shapiro based on reviews by Luca Ferretti and 2 anonymous reviewersSequencing and analyzing SARS-Cov-2 genomes in nearly real time has the potential to quickly confirm (and inform) our knowledge of, and response to, the current pandemic [1,2]. In this manuscript [3], Danesh and colleagues use the earliest set of available SARS-Cov-2 genome sequences available from France to make inferences about the timing of the major epidemic wave, the duration of infections, and the efficacy of lockdown measures. Their phylodynamic estimates -- based on fitting genomic data to molecular clock and transmission models -- are reassuringly close to estimates based on 'traditional' epidemiological methods: the French epidemic likely began in mid-January or early February 2020, and spread relatively rapidly (doubling every 3-5 days), with people remaining infectious for a median of 5 days [4,5]. These transmission parameters are broadly in line with estimates from China [6,7], but are currently unknown in France (in the absence of contact tracing data). By estimating the temporal reproductive number (Rt), the authors detected a slowing down of the epidemic in the most recent period of the study, after mid-March, supporting the efficacy of lockdown measures. References [1] Grubaugh, N. D., Ladner, J. T., Lemey, P., Pybus, O. G., Rambaut, A., Holmes, E. C., & Andersen, K. G. (2019). Tracking virus outbreaks in the twenty-first century. Nature microbiology, 4(1), 10-19. doi: 10.1038/s41564-018-0296-2 | Early phylodynamics analysis of the COVID-19 epidemics in France | Gonché Danesh, Baptiste Elie,Yannis Michalakis, Mircea T. Sofonea, Antonin Bal, Sylvie Behillil, Grégory Destras, David Boutolleau, Sonia Burrel, Anne-Geneviève Marcelin, Jean-Christophe Plantier, Vincent Thibault, Etienne Simon-Loriere, Sylvie va... | <p>France was one of the first countries to be reached by the COVID-19 pandemic. Here, we analyse 196 SARS-Cov-2 genomes collected between Jan 24 and Mar 24 2020, and perform a phylodynamics analysis. In particular, we analyse the doubling time, r... | | Evolutionary Epidemiology, Molecular Evolution, Phylogenetics / Phylogenomics | B. Jesse Shapiro | 2020-06-04 13:13:57 | ||
05 Feb 2019
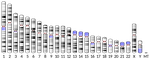
The quiescent X, the replicative Y and the AutosomesReplication-independent mutations: a universal signature ?Recommended by Nicolas Galtier based on reviews by Marc Robinson-Rechavi and Robert LanfearMutations are the primary source of genetic variation, and there is an obvious interest in characterizing and understanding the processes by which they appear. One particularly important question is the relative abundance, and nature, of replication-dependent and replication-independent mutations - the former arise as cells replicate due to DNA polymerization errors, whereas the latter are unrelated to the cell cycle. A recent experimental study in fission yeast identified a signature of mutations in quiescent (=non-replicating) cells: the spectrum of such mutations is characterized by an enrichment in insertions and deletions (indels) compared to point mutations, and an enrichment of deletions compared to insertions [2]. References [1] Achaz, G., Gangloff, S., and Arcangioli, B. (2019). The quiescent X, the replicative Y and the Autosomes. BioRxiv, 351288, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/351288 | The quiescent X, the replicative Y and the Autosomes | Guillaume Achaz, Serge Gangloff, Benoit Arcangioli | <p>From the analysis of the mutation spectrum in the 2,504 sequenced human genomes from the 1000 genomes project (phase 3), we show that sexual chromosomes (X and Y) exhibit a different proportion of indel mutations than autosomes (A), ranking the... | | Bioinformatics & Computational Biology, Genome Evolution, Human Evolution, Molecular Evolution, Population Genetics / Genomics, Reproduction and Sex | Nicolas Galtier | 2018-07-25 10:37:48 | ||
26 Nov 2019
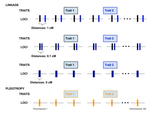
Pleiotropy or linkage? Their relative contributions to the genetic correlation of quantitative traits and detection by multi-trait GWA studiesUnderstanding the effects of linkage and pleiotropy on evolutionary adaptationRecommended by Kathleen Lotterhos based on reviews by Pär Ingvarsson and 1 anonymous reviewerGenetic correlations among traits are ubiquitous in nature. However, we still have a limited understanding of the genetic architecture of trait correlations. Some genetic correlations among traits arise because of pleiotropy - single mutations or genotypes that have effects on multiple traits. Other genetic correlations among traits arise because of linkage among mutations that have independent effects on different traits. Teasing apart the differential effects of pleiotropy and linkage on trait correlations is difficult, because they result in very similar genetic patterns. However, understanding these differential effects gives important insights into how ubiquitous pleiotropy may be in nature. References [1] Chebib, J. and Guillaume, F. (2019). Pleiotropy or linkage? Their relative contributions to the genetic correlation of quantitative traits and detection by multi-trait GWA studies. bioRxiv, 656413, v3 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/656413 | Pleiotropy or linkage? Their relative contributions to the genetic correlation of quantitative traits and detection by multi-trait GWA studies | Jobran Chebib and Frédéric Guillaume | <p>Genetic correlations between traits may cause correlated responses to selection depending on the source of those genetic dependencies. Previous models described the conditions under which genetic correlations were expected to be maintained. Sel... | | Bioinformatics & Computational Biology, Evolutionary Applications, Evolutionary Dynamics, Evolutionary Theory, Genome Evolution, Genotype-Phenotype, Molecular Evolution, Population Genetics / Genomics, Quantitative Genetics | Kathleen Lotterhos | 2019-06-05 13:51:43 | ||
20 May 2020
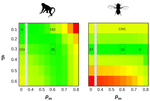
How much does Ne vary among species?Further questions on the meaning of effective population sizeRecommended by Martin Lascoux based on reviews by 3 anonymous reviewersIn spite of its name, the effective population size, Ne, has a complex and often distant relationship to census population size, as we usually understand it. In truth, it is primarily an abstract concept aimed at measuring the amount of genetic drift occurring in a population at any given time. The standard way to model random genetic drift in population genetics is the Wright-Fisher model and, with a few exceptions, definitions of the effective population size stems from it: “a certain model has effective population size, Ne, if some characteristic of the model has the same value as the corresponding characteristic for the simple Wright-Fisher model whose actual size is Ne” (Ewens 2004). Since Sewall Wright introduced the concept of effective population size in 1931 (Wright 1931), it has flourished and there are today numerous definitions of it depending on the process being examined (genetic diversity, loss of alleles, efficacy of selection) and the characteristic of the model that is considered. These different definitions of the effective population size were generally introduced to address specific aspects of the evolutionary process. One aspect that has been hotly debated since the first estimates of genetic diversity in natural populations were published is the so-called Lewontin’s paradox (1974). Lewontin noted that the observed variation in heterozygosity across species was much smaller than one would expect from the neutral expectations calculated with the actual size of the species. References Brandvain Y, Wright SI (2016) The Limits of Natural Selection in a Nonequilibrium World. Trends in Genetics, 32, 201–210. doi: 10.1016/j.tig.2016.01.004 | How much does Ne vary among species? | Nicolas Galtier, Marjolaine Rousselle | <p>Genetic drift is an important evolutionary force of strength inversely proportional to *Ne*, the effective population size. The impact of drift on genome diversity and evolution is known to vary among species, but quantifying this effect is a d... | | Bioinformatics & Computational Biology, Genome Evolution, Molecular Evolution, Population Genetics / Genomics | Martin Lascoux | 2019-12-08 00:11:00 | ||
14 Feb 2024
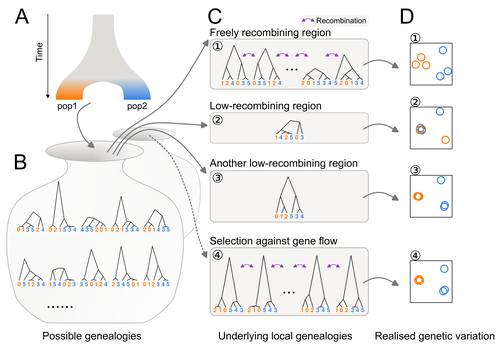
Distinct patterns of genetic variation at low-recombining genomic regions represent haplotype structureDiscerning the causes of local deviations in genetic variation: the effect of low-recombination regionsRecommended by Matteo Fumagalli based on reviews by Claire Merot and 1 anonymous reviewer based on reviews by Claire Merot and 1 anonymous reviewer
In this study, Ishigohoka and colleagues tackle an important, yet often overlooked, question on the causes of genetic variation. While genome-wide patterns represent population structure, local variation is often associated with selection. Authors propose that an alternative cause for variation in individual loci is reduced recombination rate. To test this hypothesis, authors perform local Principal Component Analysis (PCA) (Li & Ralph, 2019) to identify local deviations in population structure in the Eurasian blackcap (Sylvia atricapilla) (Ishigohoka et al. 2022). This approach is typically used to detect chromosomal rearrangements or any long region of linked loci (e.g., due to reduced recombination or selection) (Mérot et al. 2021). While other studies investigated the effect of low recombination on genetic variation (Booker et al. 2020), here authors provide a comprehensive analysis of the effect of recombination to local PCA patterns both in empirical and simulated data sets. Findings demonstrate that low recombination (and not selection) can be the sole explanatory variable for outlier windows. The study also describes patterns of genetic variation along the genome of Eurasian blackcaps, localising at least two polymorphic inversions (Ishigohoka et al. 2022). Further investigations on the effect of model parameters (e.g., window sizes and thresholds for defining low-recombining regions), as well as the use of powerful neutrality tests are in need to clearly assess whether outlier regions experience selection and reduced recombination, and to what extent. References Booker, T. R., Yeaman, S., & Whitlock, M. C. (2020). Variation in recombination rate affects detection of outliers in genome scans under neutrality. Molecular Ecology, 29 (22), 4274–4279. https://doi.org/10.1111/mec.15501 Ishigohoka, J., Bascón-Cardozo, K., Bours, A., Fuß, J., Rhie, A., Mountcastle, J., Haase, B., Chow, W., Collins, J., Howe, K., Uliano-Silva, M., Fedrigo, O., Jarvis, E. D., Pérez-Tris, J., Illera, J. C., Liedvogel, M. (2022) Distinct patterns of genetic variation at low-recombining genomic regions represent haplotype structure. bioRxiv 2021.12.22.473882, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.12.22.473882 Li, H., & Ralph, P. (2019). Local PCA Shows How the Effect of Population Structure Differs Along the Genome. Genetics, 211 (1), 289–304. https://doi.org/10.1534/genetics.118.301747 Mérot, C., Berdan, E. L., Cayuela, H., Djambazian, H., Ferchaud, A.-L., Laporte, M., Normandeau, E., Ragoussis, J., Wellenreuther, M., & Bernatchez, L. (2021). Locally Adaptive Inversions Modulate Genetic Variation at Different Geographic Scales in a Seaweed Fly. Molecular Biology and Evolution, 38 (9), 3953–3971. https://doi.org/10.1093/molbev/msab143 | Distinct patterns of genetic variation at low-recombining genomic regions represent haplotype structure | Jun Ishigohoka, Karen Bascón-Cardozo, Andrea Bours, Janina Fuß, Arang Rhie, Jacquelyn Mountcastle, Bettina Haase, William Chow, Joanna Collins, Kerstin Howe, Marcela Uliano-Silva, Olivier Fedrigo, Erich D. Jarvis, Javier Pérez-Tris, Juan Carlos Il... | <p>Genetic variation of the entire genome represents population structure, yet individual loci can show distinct patterns. Such deviations identified through genome scans have often been attributed to effects of selection instead of randomness. Th... | | Genome Evolution, Molecular Evolution, Population Genetics / Genomics | Matteo Fumagalli | 2023-10-13 11:58:47 | ||
13 Dec 2018
Separate the wheat from the chaff: genomic analysis of local adaptation in the red coral Corallium rubrumPros and Cons of local adaptation scansRecommended by Guillaume Achaz based on reviews by Lucas Gonçalves da Silva and 1 anonymous reviewerThe preprint by Pratlong et al. [1] is a well thought quest for genomic regions involved in local adaptation to depth in a species a red coral living the Mediterranean Sea. It first describes a pattern of structuration and then attempts to find candidate genes involved in local adaptation by contrasting deep with shallow populations. Although the pattern of structuration is clear and meaningful, the candidate genomic regions involved in local adaptation remain to be confirmed. Two external reviewers and myself found this preprint particularly interesting regarding the right-mindedness of the authors in front of the difficulties they encounter during their experiments. The discussions on the pros and cons of the approach are very sound and can be easily exported to a large number of studies that hunt for local adaptation. In this sense, the lessons one can learn by reading this well documented manuscript are certainly valuable for a wide range of evolutionary biologists. References [1] Pratlong, M., Haguenauer, A., Brener, K., Mitta, G., Toulza, E., Garrabou, J., Bensoussan, N., Pontarotti P., & Aurelle, D. (2018). Separate the wheat from the chaff: genomic scan for local adaptation in the red coral Corallium rubrum. bioRxiv, 306456, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/306456 | Separate the wheat from the chaff: genomic analysis of local adaptation in the red coral Corallium rubrum | Pratlong M, Haguenauer A, Brener K, Mitta G, Toulza E, Garrabou J, Bensoussan N, Pontarotti P, Aurelle D | <p>Genomic data allow an in-depth and renewed study of local adaptation. The red coral (Corallium rubrum, Cnidaria) is a highly genetically structured species and a promising model for the study of adaptive processes along an environmental gradien... | | Adaptation, Population Genetics / Genomics | Guillaume Achaz | 2018-04-24 11:27:40 | ||
23 Jan 2020

A novel workflow to improve multi-locus genotyping of wildlife species: an experimental set-up with a known model systemImproving the reliability of genotyping of multigene families in non-model organismsRecommended by François Rousset based on reviews by Sebastian Ernesto Ramos-Onsins, Helena Westerdahl and Thomas BigotThe reliability of published scientific papers has been the topic of much recent discussion, notably in the biomedical sciences [1]. Although small sample size is regularly pointed as one of the culprits, big data can also be a concern. The advent of high-throughput sequencing, and the processing of sequence data by opaque bioinformatics workflows, mean that sequences with often high error rates are produced, and that exact but slow analyses are not feasible. References [1] Ioannidis, J. P. A, Greenland, S., Hlatky, M. A., Khoury, M. J., Macleod, M. R., Moher, D., Schulz, K. F. and Tibshirani, R. (2014) Increasing value and reducing waste in research design, conduct, and analysis. The Lancet, 383, 166-175. doi: 10.1016/S0140-6736(13)62227-8 | A novel workflow to improve multi-locus genotyping of wildlife species: an experimental set-up with a known model system | Gillingham, Mark A. F., Montero, B. Karina, Wilhelm, Kerstin, Grudzus, Kara, Sommer, Simone and Santos, Pablo S. C. | <p>Genotyping novel complex multigene systems is particularly challenging in non-model organisms. Target primers frequently amplify simultaneously multiple loci leading to high PCR and sequencing artefacts such as chimeras and allele amplification... | | Bioinformatics & Computational Biology, Evolutionary Ecology, Genome Evolution, Molecular Evolution | François Rousset | Helena Westerdahl, Sebastian Ernesto Ramos-Onsins, Paul J. McMurdie , Arnaud Estoup, Vincent Segura, Jacek Radwan , Torbjørn Rognes , William Stutz , Kevin Vanneste , Thomas Bigot, Jill A. Hollenbach , Wieslaw Babik , Marie-Christin... | 2019-05-15 17:30:44 | |
23 Feb 2024
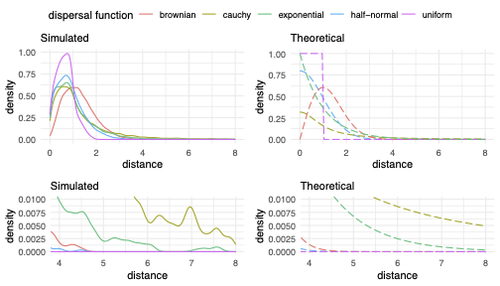
Exploring the effects of ecological parameters on the spatial structure of genetic tree sequencesDisentangling the impact of mating and competition on dispersal patternsRecommended by Diego Ortega-Del Vecchyo based on reviews by Anthony Wilder Wohns, Christian Huber and 2 anonymous reviewersSpatial population genetics is a field that studies how different evolutionary processes shape geographical patterns of genetic variation. This field is currently hampered by the lack of a deep understanding of the impact of different evolutionary processes shaping the genetic diversity observed across a continuous space (Bradburd and Ralph 2019). Luckily, the recent development of slendr (Petr et al. 2023), which uses the simulator SLiM (Haller and Messer 2023), provides a powerful tool to perform simulations to analyze the impact of different evolutionary parameters on spatial patterns of genetic variation. Here, Ianni-Ravn, Petr, and Racimo 2023 present a series of well-designed simulations to study how three evolutionary factors (dispersal distance, competition distance, and mate choice distance) shape the geographical structure of genealogies. The authors model the dispersal distance between parents and their offspring using five different distributions. Then, the authors perform simulations and they contrast the correspondence between the distribution of observed parent-offspring distances (called DD in the paper) and the distribution used in the simulations (called DF). The authors observe a reasonable correspondence between DF and DD. The authors then show that the competition distance, which decreases the fitness of individuals due to competition for resources if the individuals are close to each other, has small effects on the differences between DD and DF. In contrast, the mate choice distance (which specifies how far away can a parent go to choose a mate) causes discrepancies between DD and DF. When the mate choice distance is small, the individuals tend to cluster close to each other. Overall, these results show that the observed distances between parents and offspring are dependent on the three parameters inspected (dispersal distance, competition distance, and mate choice distance) and make the case that further ecological knowledge of each of these parameters is important to determine the processes driving the dispersal of individuals across geographical space. Based on these results, the authors argue that an “effective dispersal distance” parameter, which takes into account the impact of mate choice distance and dispersal distance, is more prone to be inferred from genetic data. The authors also assess our ability to estimate the dispersal distance using genealogical data in a scenario where the mating distance has small effects on the dispersal distance. Interestingly, the authors show that accurate estimates of the dispersal distance can be obtained when using information from all the parents and offspring going from the present back to the coalescence of all the individuals to the most recent common ancestor. On the other hand, the estimates of the dispersal distance are underestimated when less information from the parent-offspring relationships is used to estimate the dispersal distance. This paper shows the importance of considering mating patterns and the competition for resources when analyzing the dispersal of individuals. The analysis performed by the authors backs up this claim with carefully designed simulations. I recommend this preprint because it makes a strong case for the consideration of ecological factors when analyzing the structure of genealogies and the dispersal of individuals. Hopefully more studies in the future will continue to use simulations and to develop analytical theory to understand the importance of various ecological processes driving spatial genetic variation changes. Bradburd, Gideon S., and Peter L. Ralph. 2019. “Spatial Population Genetics: It’s About Time.” Annual Review of Ecology, Evolution, and Systematics 50 (1): 427–49. https://doi.org/10.1146/annurev-ecolsys-110316-022659. Haller, Benjamin C., and Philipp W. Messer. 2023. “SLiM 4: Multispecies Eco-Evolutionary Modeling.” The American Naturalist 201 (5): E127–39. https://doi.org/10.1086/723601. Ianni-Ravn, Mariadaria K., Martin Petr, and Fernando Racimo. 2023. “Exploring the Effects of Ecological Parameters on the Spatial Structure of Genealogies.” bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.03.27.534388. Petr, Martin, Benjamin C. Haller, Peter L. Ralph, and Fernando Racimo. 2023. “Slendr: A Framework for Spatio-Temporal Population Genomic Simulations on Geographic Landscapes.” Peer Community Journal 3 (e121). https://doi.org/10.24072/pcjournal.354. | Exploring the effects of ecological parameters on the spatial structure of genetic tree sequences | Mariadaria K. Ianni-Ravn, Martin Petr, Fernando Racimo | <p>Geographic space is a fundamental dimension of evolutionary change, determining how individuals disperse and interact with each other. Consequently, space has an important influence on the structure of genealogies and the distribution of geneti... | | Phylogeography & Biogeography, Population Genetics / Genomics | Diego Ortega-Del Vecchyo | 2023-03-31 18:21:02 |
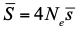 , where Ne is the effective population size and
, where Ne is the effective population size and  is the mean fitness effect of non-synonymous mutations. Assuming further that distinct species share a common DFE and therefore a common
is the mean fitness effect of non-synonymous mutations. Assuming further that distinct species share a common DFE and therefore a common  . Applying their newly developed approach to various datasets they conclude that the power of drift varies by a factor of at least 500 between large-Ne (Drosophila) and small-Ne species (H. sapiens). This is an order of magnitude larger than what would be obtained by comparing estimates of the variation in neutral diversity. Hence the proposed approach seems to have gone some way in making Lewontin’s paradox less paradoxical. But, perhaps more importantly, as the authors tersely point out at the end of the abstract their results further questions the meaning of Ne parameters in population genetics. And arguably this could well be the most important contribution of their study and something that is badly needed.
. Applying their newly developed approach to various datasets they conclude that the power of drift varies by a factor of at least 500 between large-Ne (Drosophila) and small-Ne species (H. sapiens). This is an order of magnitude larger than what would be obtained by comparing estimates of the variation in neutral diversity. Hence the proposed approach seems to have gone some way in making Lewontin’s paradox less paradoxical. But, perhaps more importantly, as the authors tersely point out at the end of the abstract their results further questions the meaning of Ne parameters in population genetics. And arguably this could well be the most important contribution of their study and something that is badly needed. 



