
Latest recommendations

| Id | Title | Authors | Abstract | Picture▲ | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
18 May 2020
The insertion of a mitochondrial selfish element into the nuclear genome and its consequencesSome evolutionary insights into an accidental homing endonuclease passage from mitochondria to the nucleusRecommended by Sylvain Charlat based on reviews by Jan Engelstaedter and Yannick WurmNot all genetic elements composing genomes are there for the benefit of their carrier. Many have no consequences on fitness, or too mild ones to be eliminated by selection, and thus stem from neutral processes. Many others are indeed the product of selection, but one acting at a different level, increasing the fitness of some elements of the genome only, at the expense of the “organism” as a whole. These can be called selfish genetic elements, and come into a wide variety of flavours [1], illustrating many possible means to cheat with “fair” reproductive processes such as meiosis, and thus get overrepresented in the offspring of their hosts. Producing copies of itself through transposition is one such strategy; a very successful one indeed, explaining a large part of the genomic content of many organisms. Killing non carrier gametes following meiosis in heterozygous carriers is another one. Less know and less common is the ability of some elements to turn heterozygous carriers into homozygous ones, that will thus transmit the selfish elements to all offspring instead of half. This is achieved by nucleic sequences encoding so-called “Homing endonucleases” (HEs). These proteins tend to induce double strand breaks of DNA specifically in regions homologous to their own insertion sites. The recombination machinery is such that the intact homologous region, that is, the one carrying the HE sequence, is then used as a template for the reparation of the break, resulting in the effective conversion of a non-carrier allele into a carrier allele. Such elements can also occur in the mitochondrial genomes of organisms where mitochondria are not strictly transmitted by one parent only, offering mitochondrial HEs some opportunities for “homing” into new non carrier genomes. This is the case in yeasts, where HEs were first reported [2,3]. References [1] Burt, A., and Trivers, R. (2006). Genes in Conflict: The Biology of Selfish Genetic Elements. Belknap Press. | The insertion of a mitochondrial selfish element into the nuclear genome and its consequences | Julien Y. Dutheil, Karin Münch, Klaas Schotanus, Eva H. Stukenbrock and Regine Kahmann | <p>Homing endonucleases (HE) are enzymes capable of cutting DNA at highly specific target sequences, the repair of the generated double-strand break resulting in the insertion of the HE-encoding gene ("homing" mechanism). HEs are present in all th... | | Genome Evolution, Molecular Evolution | Sylvain Charlat | 2019-09-30 20:34:23 | ||
31 Mar 2017

POSTPRINT
Human adaptation of Ebola virus during the West African outbreakEbola evolution during the 2013-2016 outbreakRecommended by Sylvain Gandon and Sébastien LionThe Ebola virus (EBOV) epidemic that started in December 2013 resulted in around 28,000 cases and more than 11,000 deaths. Since the emergence of the disease in Zaire in 1976 the virus had produced a number of outbreaks in Africa but until 2013 the reported numbers of human cases had never risen above 500. Could this exceptional epidemic size be due to the spread of a human-adapted form of the virus? The large mutation rate of the virus [1-2] may indeed introduce massive amounts of genetic variation upon which selection may act. Several earlier studies based on the accumulation of genome sequences sampled during the epidemic led to contrasting conclusions. A few studies discussed evidence of positive selection on the glycoprotein that may be linked to phenotypic variations on infectivity and/or immune evasion [3-4]. But the heterogeneity in the transmission of some lineages could also be due to environmental heterogeneity and/or stochasticity. Most studies could not rule out the null hypothesis of the absence of positive selection and human adaptation [1-2 and 5]. In a recent experimental study, Urbanowicz et al. [6] chose a different method to tackle this question. A phylogenetic analysis of genome sequences from viruses sampled in West Africa revealed the existence of two main lineages (one with a narrow geographic distribution in Guinea, and the other with a wider geographic distribution) distinguished by a single amino acid substitution in the glycoprotein of the virus (A82V), and of several sub-lineages characterised by additional substitutions. The authors used this phylogenetic data to generate a panel of mutant pseudoviruses and to test their ability to infect human and fruit bat cells. These experiments revealed that specific amino acid substitutions led to higher infectivity of human cells, including A82V. This increased infectivity on human cells was associated with a decreased infectivity in fruit bat cell cultures. Since fruit bats are likely to be the reservoir of the virus, this paper indicates that human adaptation may have led to a specialization of the virus to a new host. An accompanying paper in the same issue of Cell by Diehl et al. [7] reports results that confirm the trend identified by Urbanowicz et al. [6] and further indicate that the increased infectivity of A82V is specific for primate cells. Diehl et al. [7] also report some evidence for higher virulence of A82V in humans. In other words, the evolution of the virus may have led to higher abilities to infect and to kill its novel host. This work thus confirms the adaptive potential of RNA virus and the ability of Ebola to specialize to a novel host. In this context, the availability of an effective vaccine against the disease is particularly welcome [8]. The study of Urbanowicz et al. [6] is also remarkable because it illustrates the need of experimental approaches for the study of phenotypic variation when inference methods based on phylodynamics fail to extract a clear biological message. The analysis of genomic evolution is still in its infancy and there is a need for new theoretical developments to help detect more rapidly candidate mutations involved in adaptations to new environmental conditions. References [1] Gire, S.K., Goba, A., Andersen, K.G., Sealfon, R.S.G., Park, D.J., Kanneh, L., Jalloh, S., Momoh, M., Fullah, M., Dudas, G., et al. (2014). Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345, 1369–1372. doi: 10.1126/science.1259657 | Human adaptation of Ebola virus during the West African outbreak | Urbanowicz, R.A., McClure, C.P., Sakuntabhai, A., Sall, A.A., Kobinger, G., Müller, M.A., Holmes, E.C., Rey, F.A., Simon-Loriere, E., and Ball, J.K. | The 2013–2016 outbreak of Ebola virus (EBOV) in West Africa was the largest recorded. It began following the cross-species transmission of EBOV from an animal reservoir, most likely bats, into humans, with phylogenetic analysis revealing the co-ci... | | Adaptation, Evolutionary Epidemiology, Genome Evolution, Genotype-Phenotype, Molecular Evolution, Species interactions | Sylvain Gandon | 2017-03-31 14:20:38 | ||
27 Jul 2020
Evolution of the DAN gene family in vertebratesAn evolutionary view of a biomedically important gene familyRecommended by Kateryna Makova based on reviews by 2 anonymous reviewersThis manuscript [1] investigates the evolutionary history of the DAN gene family—a group of genes important for embryonic development of limbs, kidneys, and left-right axis speciation. This gene family has also been implicated in a number of diseases, including cancer and nephropathies. DAN genes have been associated with the inhibition of the bone morphogenetic protein (BMP) signaling pathway. Despite this detailed biochemical and functional knowledge and clear importance for development and disease, evolution of this gene family has remained understudied. The diversification of this gene family was investigated in all major groups of vertebrates. The monophyly of the gene members belonging to this gene family was confirmed. A total of five clades were delineated, and two novel lineages were discovered. The first lineage was only retained in cephalochordates (amphioxus), whereas the second one (GREM3) was retained by cartilaginous fish, holostean fish, and coelanth. Moreover, the patterns of chromosomal synteny in the chromosomal regions harboring DAN genes were investigated. Additionally, the authors reconstructed the ancestral gene repertoires and studied the differential retention/loss of individual gene members across the phylogeny. They concluded that the ancestor of gnathostome vertebrates possessed eight DAN genes that underwent differential retention during the evolutionary history of this group. During radiation of vertebrates, GREM1, GREM2, SOST, SOSTDC1, and NBL1 were retained in all major vertebrate groups. At the same time, GREM3, CER1, and DAND5 were differentially lost in some vertebrate lineages. At least two DAN genes were present in the common ancestor of vertebrates, and at least three DAN genes were present in the common ancestor of chordates. Therefore the patterns of retention and diversification in this gene family appear to be complex. Evolutionary slowdown for the DAN gene family was observed in mammals, suggesting selective constraints. Overall, this article puts the biomedical importance of the DAN family in the evolutionary perspective. References [1] Opazo JC, Hoffmann FG, Zavala K, Edwards SV (2020) Evolution of the DAN gene family in vertebrates. bioRxiv, 794404, ver. 3 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/794404 | Evolution of the DAN gene family in vertebrates | Juan C. Opazo, Federico G. Hoffmann, Kattina Zavala, Scott V. Edwards | <p>The DAN gene family (DAN, Differential screening-selected gene Aberrant in Neuroblastoma) is a group of genes that is expressed during development and plays fundamental roles in limb bud formation and digitation, kidney formation and morphogene... | | Molecular Evolution | Kateryna Makova | 2019-10-15 16:43:13 | ||
29 Jul 2020

POSTPRINT
The Y chromosome may contribute to sex-specific ageing in DrosophilaY chromosome makes fruit flies die youngerRecommended by Gabriel Marais, Jean-François Lemaitre and Cristina VieiraIn most animal species, males and females display distinct survival prospect, a phenomenon known as sex gap in longevity (SGL, Marais et al. 2018). The study of SGLs is crucial not only for having a full picture of the causes underlying organisms’ health, aging and death but also to initiate the development of sex-specific anti-aging interventions in humans (Austad and Bartke 2015). Three non-mutually evolutionary causes have been proposed to underlie SGLs (Marais et al. 2018). First, SGLs could be the consequences of sex-differences in life history strategies. For example, evolving dimorphic traits (e.g. body size, ornaments or armaments) may imply unequal physiological costs (e.g. developmental, maintenance) between the sexes and this may result in differences in longevity and aging. Second, mitochondria are usually transmitted by the mother and thus selection is blind to mitochondrial deleterious mutations affecting only males. Such mutations can freely accumulate in the mitochondrial genome and may reduce male longevity, a phenomenon called the mother’s curse (Frank and Hurst 1996). Third, in species with sex chromosomes, all recessive deleterious mutations will be expressed on the single X chromosome in XY males and may reduce their longevity (the unguarded X effect). In addition, the numerous transposable elements (TEs) on the Y chromosome may affect aging. TE activity is normally repressed by epigenetic regulation (DNA methylation, histone modifications and small RNAs). However, it is known that this regulation is disrupted with increasing age. Because of the TE-rich Y chromosome, more TEs may become active in old males than in old females, generating more somatic mutations, accelerating aging and reducing longevity in males (the toxic Y effect, Marais et al. 2018). References Austad, S. N., and Bartke, A. (2015). Sex differences in longevity and in responses to anti-aging interventions: A Mini-Review. Gerontology, 62(1), 40–46. 10.1159/000381472 | The Y chromosome may contribute to sex-specific ageing in Drosophila | Emily J Brown, Alison H Nguyen, Doris Bachtrog | <p>Heterochromatin suppresses repetitive DNA, and a loss of heterochromatin has been observed in aged cells of several species, including humans and *Drosophila*. Males often contain substantially more heterochromatic DNA than females, due to the ... | | Bioinformatics & Computational Biology, Expression Studies, Genetic conflicts, Genome Evolution, Genotype-Phenotype, Molecular Evolution, Reproduction and Sex | Gabriel Marais | 2020-07-28 15:06:18 | ||
14 Dec 2016

POSTPRINT
The Red Queen lives: epistasis between linked resistance lociEvidence of epistasis provides further support to the Red Queen theory of host-parasite coevolutionRecommended by Adele Mennerat and Thierry LefèvreAccording to the Red Queen theory of antagonistic host-parasite coevolution, adaptation of parasites to the most common host genotype results in negative frequency-dependent selection whereby rare host genotypes are favoured. Assuming that host resistance relies on a genetic host-parasite (mis)match involving several linked loci, then recombination appears as much more efficient than parthenogenesis in generating new resistant host genotypes. This has long been proposed to explain one of the biggest so-called paradoxes in evolutionary biology, i.e. the maintenance of recombination despite its twofold cost. Evidence from various study systems indicates that successful infection (and hence host resistance) depends on a genetic match between the parasite’s and the host’s genotype via molecular interactions involving elicitor/receptor mechanisms. However the key assumption of epistasis, i.e. that this genetic host-parasite match involves several linked resistance loci, remained unsupported so far. Metzger and coauthors [1] now provide empirical support for it. Daphnia magna can reproduce both sexually and clonally and their well-studied interaction with Pasteuria ramosa makes them an excellent model system to investigate the genetics of host resistance. D. magna hosts were found to be either resistant (complete lack of attachment of parasite spores to the host’s foregut) or susceptible (full attachment). In this study the authors carried out an elegant Mendelian genetic investigation by performing multiple crosses between four host genotypes differing in their resistance to two different parasite isolates [1]. Their results show that resistance of D. magna to each of the two P. ramosa isolates relies on Mendelian inheritance at two loci that are linked (A and B), each of them having two alleles with dominant resistance; furthermore resistance to one parasite isolate confers susceptibility to the other. They also show that a third locus appears to confer double resistance (C), but that even double resistant hosts remain susceptible to other parasite isolates, and hence that universal host resistance is lacking – all of this supporting the Red Queen theory. This paper demonstrates with a high level of clarity that host resistance is governed by multiple linked loci. The assumption of epistasis between resistance loci is supported, which makes it possible for sexual recombination to be maintained by antagonistic host-parasite coevolution. Reference [1] Metzger CMJA, Luijckx P, Bento G, Mariadassou M, Ebert D. 2016. The Red Queen lives: epistasis between linked resistance loci. Evolution 70:480-487. doi: 10.1111/evo.12854 | The Red Queen lives: epistasis between linked resistance loci | Metzger CMJA, Luijckx P, Bento G, Mariadassou M, Ebert D. | A popular theory explaining the maintenance of genetic recombination (sex) is the Red Queen Theory. This theory revolves around the idea that time-lagged negative frequency-dependent selection by parasites favors rare host genotypes generated thro... | | Evolutionary Dynamics, Evolutionary Theory, Reproduction and Sex, Species interactions | Adele Mennerat | 2016-12-14 13:58:53 | ||
02 Nov 2022
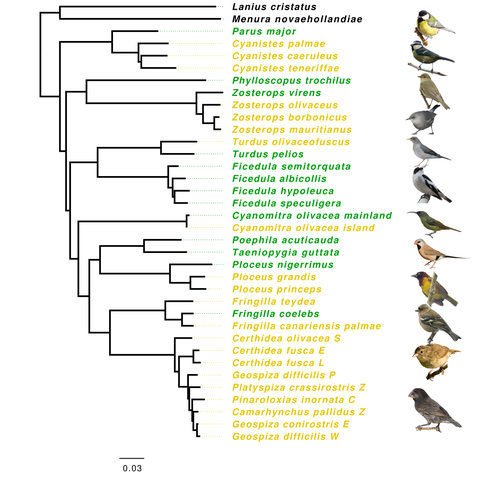
Evolution of immune genes in island birds: reduction in population sizes can explain island syndromeDemographic effects may affect adaptation to islandsRecommended by Emma Berdan based on reviews by Steven Fiddaman and 3 anonymous reviewersThe unique challenges associated with living on an island often result in organisms displaying a specific suite of traits commonly referred to as “island syndrome” (Adler and Levins, 1994; Burns, 2019; Baeckens and Van Damme, 2020). Large phenotypic shifts such as changes in size (e.g. shifts to gigantism or dwarfism, Lomolino, 2005) or coloration (Doutrelant et al., 2016) abound in the literature. However, less obvious phenotypes may also play a key role in adaptation to islands. One such trait, reduced immune function, has important implications for the future of island populations in the face of anthropogenic-induced changes. Due to lower parasite pressure caused by a less diverse and less virulent parasite population, island hosts may show a decrease in immune defenses (Beadell et al., 2006; Pérez‐Rodríguez et al., 2013). However, this hypothesis has been challenged, as many studies have found ambiguous or conflicting results (Matson, 2006; Illera et al., 2015). While most previous work has examined various immunological parameters (e.g., antibody concentrations), here, Barthe et al. (2022) take the novel approach of examining molecular signatures of immune genes. Using comparative genomic data from 34 different species of birds the authors examine the ratio of synonymous substitutions (i.e., not changing an amino acid) to non-synonymous substitutions (i.e., changing an amino acid) in innate and acquired immune genes (Pn/Ps ratio). Because population sizes on islands are lower which will affect molecular evolution, they compare these results to data from 97 control genes. Assuming relaxed selection on islands predicts that the difference between the Pn/Ps ratio of immune genes and of control genes (ΔPn/Ps) is greater in island species compared to mainland ones. As with previous work the authors found that the results differ depending on the category of immune genes. Both forms of innate defense: beta-defensins and Toll-like receptors did not show higher ΔPn/Ps for island populations. As these genes still have a higher Pn/Ps than control genes, the authors argue these results are in line with these genes being under purifying selection but lacking an “island effect”. Instead, the authors argue that demographic effects (i.e., reductions in Ne) may lead to the decreased immunity documented in other studies. In contrast, there was a reduction in Pn/Ps in MHC II genes, known to be under balancing selection. This reduction was stronger in island species and thus the authors argue that this is the only class of genes where a role for relaxed selection can be invoked. Together these results demonstrate that the changes in immunity experienced by island species are complex and that different categories of immune genes can experience different selective pressures. By including control genes in their study, they particularly highlight the importance of accounting for shifts in Ne when examining patterns of island species evolution. Hopefully, this kind of framework will be applied to other taxa to determine if these results are widespread or more specific to birds. References Adler GH, Levins R (1994) The Island Syndrome in Rodent Populations. The Quarterly Review of Biology, 69, 473–490. https://doi.org/10.1086/418744 Baeckens S, Van Damme R (2020) The island syndrome. Current Biology, 30, R338–R339. https://doi.org/10.1016/j.cub.2020.03.029 Barthe M, Doutrelant C, Covas R, Melo M, Illera JC, Tilak M-K, Colombier C, Leroy T, Loiseau C, Nabholz B (2022) Evolution of immune genes in island birds: reduction in population sizes can explain island syndrome. bioRxiv, 2021.11.21.469450, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.11.21.469450 Beadell JS, Ishtiaq F, Covas R, Melo M, Warren BH, Atkinson CT, Bensch S, Graves GR, Jhala YV, Peirce MA, Rahmani AR, Fonseca DM, Fleischer RC (2006) Global phylogeographic limits of Hawaii’s avian malaria. Proceedings of the Royal Society B: Biological Sciences, 273, 2935–2944. https://doi.org/10.1098/rspb.2006.3671 Burns KC (2019) Evolution in Isolation: The Search for an Island Syndrome in Plants. Cambridge University Press, Cambridge. https://doi.org/10.1017/9781108379953 Doutrelant C, Paquet M, Renoult JP, Grégoire A, Crochet P-A, Covas R (2016) Worldwide patterns of bird colouration on islands. Ecology Letters, 19, 537–545. https://doi.org/10.1111/ele.12588 Illera JC, Fernández-Álvarez Á, Hernández-Flores CN, Foronda P (2015) Unforeseen biogeographical patterns in a multiple parasite system in Macaronesia. Journal of Biogeography, 42, 1858–1870. https://doi.org/10.1111/jbi.12548 Lomolino MV (2005) Body size evolution in insular vertebrates: generality of the island rule. Journal of Biogeography, 32, 1683–1699. https://doi.org/10.1111/j.1365-2699.2005.01314.x Matson KD (2006) Are there differences in immune function between continental and insular birds? Proceedings of the Royal Society B: Biological Sciences, 273, 2267–2274. https://doi.org/10.1098/rspb.2006.3590 Pérez-Rodríguez A, Ramírez Á, Richardson DS, Pérez-Tris J (2013) Evolution of parasite island syndromes without long-term host population isolation: parasite dynamics in Macaronesian blackcaps Sylvia atricapilla. Global Ecology and Biogeography, 22, 1272–1281. https://doi.org/10.1111/geb.12084 | Evolution of immune genes in island birds: reduction in population sizes can explain island syndrome | Mathilde BARTHE, Claire DOUTRELANT, Rita COVAS, Martim MELO, Juan Carlos ILLERA, Marie-Ka TILAK, Constance COLOMBIER, Thibault LEROY , Claire LOISEAU , Benoit NABHOLZ | <p style="text-align: justify;">Shared ecological conditions encountered by species that colonize islands often lead to the evolution of convergent phenotypes, commonly referred to as “island syndrome”. Reduced immune functions have been previousl... | | Adaptation, Molecular Evolution, Population Genetics / Genomics | Emma Berdan | 2021-11-28 11:01:31 | ||
12 Nov 2021

How ancient forest fragmentation and riparian connectivity generate high levels of genetic diversity in a micro-endemic Malagasy treeAn ancient age of open-canopy landscapes in northern Madagascar? Evidence from the population genetic structure of a forest treeRecommended by Miguel de Navascués based on reviews by Katharina Budde and Yurena Arjona based on reviews by Katharina Budde and Yurena Arjona
We currently live in the Anthropocene, the geological age characterized by a profound impact of human populations in the ecosystems and the environment. While there is little doubt about the action of humans in the shaping of present landscapes, it can be difficult to determine what the state of those landscapes was before humans started to modify them. This is the case of the Madagascar grasslands, whose origins have been debated with arguments proposing them either as anthropogenic, created with the arrival of humans around 2000BP, or as ancient features of the natural landscape with a forest fragmentation process due to environmental changes pre-dating human arrival [e.g. 1,2]. One way to clarify this question is through the genetic study of native species. Population continuity and fragmentation along time shape the structure of the genetic diversity in space. Species living in a uniform continuous habitat are expected to show genetic structuring determined only by geographical distance. Recent changes of the habitat can take many generations to reshape that genetic structure [3]. Thus, we expect genetic structure to reflect ancient features of the landscape. The work by Jordi Salmona and collaborators [4] studies the factors determining the population genetic structure of the Malagasy spiny olive (Noronhia spinifolia). This narrow endemic species is distributed in the discontinuous forest patches of the Loky-Manambato region (northern Madagascar). Jordi Salmona and collaborators genotyped 72 individuals distributed across the species distribution with restriction associated DNA sequencing and organelle microsatellite markers. Then, they studied the population genetic structure of the species. Using isolation-by-resistance models [5], they tested the influence of several landscape features (forest cover, roads, rivers, slope, etc.) on the connectivity between populations. Maternally inherited loci (chloroplast and mitochondria) and bi-parentally inherited loci (nuclear), were analysed separately in an attempt to identify the role of pollen and seed dispersal in the connectivity of populations. Despite the small distribution of the species, Jordi Salmona and collaborators [4] found remarkable levels of genetic diversity. The spatial structure of this diversity was found to be mainly explained by the forest cover of the landscape, suggesting that the landscape has been composed by patches of forests and grasslands for a long time. The main role of forest cover for the connectivity among populations also highlights the importance of riparian forest as dispersal corridors. Finally, differences between organelle and nuclear markers were not enough to establish any strong conclusion about the differences between pollen and seed dispersal. The results presented by Jordi Salmona and collaborators [4] contribute to the understanding of the history and ecology of understudied Madagascar ecosystems. Previous population genetic studies in some forest-dwelling mammals have been interpreted as supporting an old age for the fragmented landscapes in northern Madagascar [e.g. 1,6]. To my knowledge, this is the first study on a tree species. While this work might not completely settle the debate, it emphasizes the importance of studying a diversity of species to understand the biogeographic dynamics of a region. References 1. Quéméré, E., X. Amelot, J. Pierson, B. Crouau-Roy, L. Chikhi (2012) Genetic data suggest a natural prehuman origin of open habitats in northern Madagascar and question the deforestation narrative in this region. Proceedings of the National Academy of Sciences of the United States of | How ancient forest fragmentation and riparian connectivity generate high levels of genetic diversity in a micro-endemic Malagasy tree | Jordi Salmona, Axel Dresen, Anicet E. Ranaivoson, Sophie Manzi, Barbara Le Pors, Cynthia Hong-Wa, Jacqueline Razanatsoa, Nicole V. Andriaholinirina, Solofonirina Rasoloharijaona, Marie-Elodie Vavitsara, Guillaume Besnard | <p>Understanding landscape changes is central to predicting evolutionary trajectories and defining conservation practices. While human-driven deforestation is intense throughout Madagascar, exception in areas like the Loky-Manambato region (North)... | | Evolutionary Ecology, Phylogeography & Biogeography, Population Genetics / Genomics | Miguel de Navascués | 2020-11-27 09:07:21 | ||
24 Oct 2022

Evolutionary responses of energy metabolism, development, and reproduction to artificial selection for increasing heat tolerance in Drosophila subobscuraThe other side of the evolution of heat tolerance: correlated responses in metabolism and life-history traitsRecommended by Inês Fragata and Pedro Simões based on reviews by Marija Savić Veselinović and 1 anonymous reviewer
Understanding how species respond to environmental changes is becoming increasingly important in order to predict the future of biodiversity and species distributions under current global warming conditions (Rezende 2020; Bennett et al 2021). Two key factors to take into account in these predictions are the tolerance of organisms to heat stress and subsequently how they adapt to increasingly warmer temperatures. Coupled with this, one important factor that is often overlooked when addressing the evolution of thermal tolerance, is the correlated responses in traits that are important to fitness, such as life histories, behavior and the underlying metabolic processes. The rate and intensity of the thermal stress are expected to be major factors in shaping the evolution of heat tolerance and correlated responses in other traits. For instance, lower rates of thermal stress are predicted to select for individuals with a slower metabolism (Santos et al 2012), whereas low metabolism is expected to lead to a lower reproductive rate (Dammhahn et al 2018). To quantify the importance of the rate and intensity of thermal stress on the evolutionary response of heat tolerance and correlated response in behavior, Mesas et al (2021) performed experimental evolution in Drosophila subobscura using selective regimes with slow or fast ramping protocols. Whereas both regimes showed increased heat tolerance with similar evolutionary rates, the correlated responses in thermal performance curves for locomotor behavior differed between selection regimes. These findings suggest that thermal rate and intensity may shape the evolution of correlated responses in other traits, urging the need to understand possible correlated responses at relevant levels such as life history and metabolism. In the present contribution, Mesas and Castañeda (2022) investigate whether the disparity in thermal performance curves observed in the previous experiment (Mesas et al 2021) could be explained by differences in metabolic energy production and consumption, and how this correlated with the reproductive output (fecundity and viability). Overall, the authors show some evidence for lowered enzyme activity and increased performance in life-history traits, particularly for the slow-ramping selected flies. Specifically, the authors observe a reduction in glucose metabolism and increased viability when evolving under slow ramping stress. Interestingly, both regimes show a general increase in fecundity, suggesting that adaptation to these higher temperatures is not costly (for reproduction) in the ancestral environment. The evidence for a somewhat lower metabolism in the slow-ramping lines suggests the evolution of a slow “pace of life”. The “pace of life” concept tries to bridge variation across several levels namely metabolism, physiology, behavior and life history, with low “pace of life” organisms presenting lower metabolic rates, later reproduction and higher longevity than fast “pace of life” organisms (Dammhahn et al 2018, Tuzun & Stocks 2022). As the authors state there is not a clear-cut association with the expectations of the pace of life hypothesis since there was evidence for increased reproductive output under both selection intensity regimes. This suggests that, given sufficient trait genetic variance, positively correlated responses may emerge during some stages of thermal evolution. As fecundity estimates in this study were focussed on early life, the possibility of a decrease in the cumulative reproductive output of the selected flies, even under benign conditions, cannot be excluded. This would help explain the apparent paradox of increased fecundity in selected lines. In this context, it would also be interesting to explore the variation in reproductive output at different temperatures, i.e to obtain thermal performance curves for life histories. Mesas and Castañeda (2022) raise important questions to pursue in the future and contribute to the growing evidence that, in order to predict the distribution of ectothermic species under current global warming conditions, we need to expand beyond determining the physiological thermal limits of each organism (Parratt et al 2021). Ultimately, integrating metabolic, life-history and behavioral changes during evolution under different thermal stresses within a coherent framework is key to developing better predictions of temperature effects on natural populations. Bennett, J.M., Sunday, J., Calosi, P. et al. The evolution of critical thermal limits of life on Earth. Nat Commun 12, 1198 (2021). https://doi.org/10.1038/s41467-021-21263-8 Dammhahn, M., Dingemanse, N.J., Niemelä, P.T. et al. Pace-of-life syndromes: a framework for the adaptive integration of behaviour, physiology and life history. Behav Ecol Sociobiol 72, 62 (2018). https://doi.org/10.1007/s00265-018-2473-y Mesas, A, Jaramillo, A, Castañeda, LE. Experimental evolution on heat tolerance and thermal performance curves under contrasting thermal selection in Drosophila subobscura. J Evol Biol 34, 767– 778 (2021). https://doi.org/10.1111/jeb.13777 Parratt, S.R., Walsh, B.S., Metelmann, S. et al. Temperatures that sterilize males better match global species distributions than lethal temperatures. Nat. Clim. Chang. 11, 481–484 (2021). https://doi.org/10.1038/s41558-021-01047-0 Santos, M, Castañeda, LE, Rezende, EL Keeping pace with climate change: what is wrong with the evolutionary potential of upper thermal limits? Ecology and evolution, 2(11), 2866-2880 (2012). https://doi.org/10.1002/ece3.385 Tüzün, N, Stoks, R. A fast pace-of-life is traded off against a high thermal performance. Proceedings of the Royal Society B, 289(1972), 20212414 (2022). https://doi.org/10.1098/rspb.2021.2414 Rezende, EL, Bozinovic, F, Szilágyi, A, Santos, M. Predicting temperature mortality and selection in natural Drosophila populations. Science, 369(6508), 1242-1245 (2020). https://doi.org/10.1126/science.aba9287 | Evolutionary responses of energy metabolism, development, and reproduction to artificial selection for increasing heat tolerance in Drosophila subobscura | Andres Mesas, Luis E. Castaneda | <p>Adaptations to warming conditions exhibited by ectotherms include increasing heat tolerance but also metabolic changes to reduce maintenance costs (metabolic depression), which can allow them to redistribute the energy surplus to biological fun... | | Adaptation, Evolutionary Ecology, Experimental Evolution, Life History | Inês Fragata | 2022-02-08 01:05:50 | ||
29 Nov 2022

Joint inference of adaptive and demographic history from temporal population genomic dataInference of genome-wide processes using temporal population genomic dataRecommended by Aurelien Tellier based on reviews by Lawrence Uricchio and 2 anonymous reviewersEvolutionary genomics, and population genetics in particular, aim to decipher the respective influence of neutral and selective forces shaping genetic polymorphism in a species/population. This is a much-needed requirement before scanning genome data for footprints of species adaptation to their biotic and abiotic environment (Johri et al. 2022). In general, we would like to quantify the proportion of the genome evolving neutrally and under selective (positive, balancing and negative) pressures (Kern and Hahn 2018, Johri et al. 2021). We thus need to understand patterns of linked selection along the genome, that is how the distribution of genetic polymorphisms is shaped by selected sites and the recombination landscape. The present contribution by Pavinato et al. (2022) provides an additional method in the population genomics toolbox to quantify the extent of linked positive and negative selection using temporal data. The availability of genomics data for model and non-model species has led to improvement of the modeling framework for demography and selection (Johri et al. 2022), but also new inference methods making use of the full genome data based on the Sequential Markovian Coalescent (SMC, Li and Durbin 2011), Approximate Bayesian Computation (ABC, Jay et al. 2019), ABC and machine learning (Pudlo et al. 2016, Raynal et al. 2019) or Deep Learning (Sanchez et al. 2021). These methods are based on one sample in time and the use of the coalescent theory to reconstruct the past (demographic) history. However, it is also possible to obtain for many species temporal data sampled over several time points. For species with short generation time (in experimental evolution or monitored populations), one can sample a population every couple of generations as exemplified with Drosophila melanogaster (Bergland et al. 2010). For species with longer generation times that cannot be easily regularly sampled in time, it becomes possible to sequence available specimens from museums (e.g. Cridland et al. 2018) or ancient DNA samples. Methods using temporal data are based on the classical population genomics assumption that demography (migration, population subdivision, population size changes) leaves a genome-wide signal, while selection leaves a localized signal in the close vicinity of the causal mutation. Several methods do assess the demography of a population (change in effective population size, Ne, in time) using temporal data (e.g. Jorde and Ryman 2007) which can be used to calibrate the detection of loci under strong positive selection (Foll et al. 2014). Recently Buffalo and Coop (2020) used genome-wide covariance between allele frequency changes across time samples (and across replicates) to quantify the effects of linked selection over short timescales. In the present contribution, Pavinato et al. (2022) make use of temporal data to draw the joint estimation of demographic and selective parameters using a simulation-based method (ABC-Random Forests). This study by Pavinato et al. (2022) builds a framework allowing to infer the census size of the population in time (N) separately from the effect of genetic drift, which is determined by change in effective population size (Ne) in time, as well estimates of genome-wide parameters of selection. In a nutshell, the authors use a forward simulator and summarize genome data by genomic windows using classic statistics (nucleotide diversity, Tajima’s D, FST, heterozygosity) between time samples and for each sample. They specifically use the distributions (higher moments) of these statistics among all windows. The authors combine as input for the ABC-RF, vectors of summary statistics, model parameters and five latent variables: Ne, the ratio Ne/N, the number of beneficial mutations under strong selection, the average selection coefficient of strongly selected mutations, and the average substitution load. Indeed, the authors are interested in three different types of selection components: 1) the adaptive potential of a population which is estimated as the population mutation rate of beneficial mutations (θb), 2) the number of mutations under strong selection (irrespective of whether they reached fixation or not), and 3) the overall population fitness which is a function of the genetic load. In other words, the novelty of this method is not to focus on the detection of loci under selection, but to infer key parameters/distributions summarizing the genome-wide signal of demography and (positive and negative) selection. As a proof of principle, the authors then apply their method to a dataset of feral populations of honey bees (Apis mellifera) collected in California across many years and recovered from Museum samples (Cridland et al. 2018). The approach yields estimates of Ne which are on the same order of magnitude of previous estimates in hymenopterans, and the authors discuss why the different populations show various values of Ne and N which can be explained by different history of admixture with wild but also domesticated lineages of bees. This study focuses on quantifying the genome-wide joint footprints of demography, and strong positive and negative selection to determine which proportion of the genome evolves neutrally or not. Further application of this method can be anticipated, for example, to study species with ecological and life-history traits which generate discrepancies between census size and Ne, for example for plants with selfing or seed banking (Sellinger et al. 2020), and for which the genome-wide effect of linked selection is not fully understood. References Johri P, Aquadro CF, Beaumont M, Charlesworth B, Excoffier L, Eyre-Walker A, Keightley PD, Lynch M, McVean G, Payseur BA, Pfeifer SP, Stephan W, Jensen JD (2022) Recommendations for improving statistical inference in population genomics. PLOS Biology, 20, e3001669. https://doi.org/10.1371/journal.pbio.3001669 Kern AD, Hahn MW (2018) The Neutral Theory in Light of Natural Selection. Molecular Biology and Evolution, 35, 1366–1371. https://doi.org/10.1093/molbev/msy092 Johri P, Riall K, Becher H, Excoffier L, Charlesworth B, Jensen JD (2021) The Impact of Purifying and Background Selection on the Inference of Population History: Problems and Prospects. Molecular Biology and Evolution, 38, 2986–3003. https://doi.org/10.1093/molbev/msab050 Pavinato VAC, Mita SD, Marin J-M, Navascués M de (2022) Joint inference of adaptive and demographic history from temporal population genomic data. bioRxiv, 2021.03.12.435133, ver. 6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.03.12.435133 Li H, Durbin R (2011) Inference of human population history from individual whole-genome sequences. Nature, 475, 493–496. https://doi.org/10.1038/nature10231 Jay F, Boitard S, Austerlitz F (2019) An ABC Method for Whole-Genome Sequence Data: Inferring Paleolithic and Neolithic Human Expansions. Molecular Biology and Evolution, 36, 1565–1579. https://doi.org/10.1093/molbev/msz038 Pudlo P, Marin J-M, Estoup A, Cornuet J-M, Gautier M, Robert CP (2016) Reliable ABC model choice via random forests. Bioinformatics, 32, 859–866. https://doi.org/10.1093/bioinformatics/btv684 Raynal L, Marin J-M, Pudlo P, Ribatet M, Robert CP, Estoup A (2019) ABC random forests for Bayesian parameter inference. Bioinformatics, 35, 1720–1728. https://doi.org/10.1093/bioinformatics/bty867 Sanchez T, Cury J, Charpiat G, Jay F (2021) Deep learning for population size history inference: Design, comparison and combination with approximate Bayesian computation. Molecular Ecology Resources, 21, 2645–2660. https://doi.org/10.1111/1755-0998.13224 Bergland AO, Behrman EL, O’Brien KR, Schmidt PS, Petrov DA (2014) Genomic Evidence of Rapid and Stable Adaptive Oscillations over Seasonal Time Scales in Drosophila. PLOS Genetics, 10, e1004775. https://doi.org/10.1371/journal.pgen.1004775 Cridland JM, Ramirez SR, Dean CA, Sciligo A, Tsutsui ND (2018) Genome Sequencing of Museum Specimens Reveals Rapid Changes in the Genetic Composition of Honey Bees in California. Genome Biology and Evolution, 10, 458–472. https://doi.org/10.1093/gbe/evy007 Jorde PE, Ryman N (2007) Unbiased Estimator for Genetic Drift and Effective Population Size. Genetics, 177, 927–935. https://doi.org/10.1534/genetics.107.075481 Foll M, Shim H, Jensen JD (2015) WFABC: a Wright–Fisher ABC-based approach for inferring effective population sizes and selection coefficients from time-sampled data. Molecular Ecology Resources, 15, 87–98. https://doi.org/10.1111/1755-0998.12280 Buffalo V, Coop G (2020) Estimating the genome-wide contribution of selection to temporal allele frequency change. Proceedings of the National Academy of Sciences, 117, 20672–20680. https://doi.org/10.1073/pnas.1919039117 Sellinger TPP, Awad DA, Moest M, Tellier A (2020) Inference of past demography, dormancy and self-fertilization rates from whole genome sequence data. PLOS Genetics, 16, e1008698. https://doi.org/10.1371/journal.pgen.1008698 | Joint inference of adaptive and demographic history from temporal population genomic data | Vitor A. C. Pavinato, Stéphane De Mita, Jean-Michel Marin, Miguel de Navascués | <p style="text-align: justify;">Disentangling the effects of selection and drift is a long-standing problem in population genetics. Simulations show that pervasive selection may bias the inference of demography. Ideally, models for the inference o... | | Adaptation, Population Genetics / Genomics | Aurelien Tellier | 2021-10-20 09:41:26 | ||
30 Mar 2023
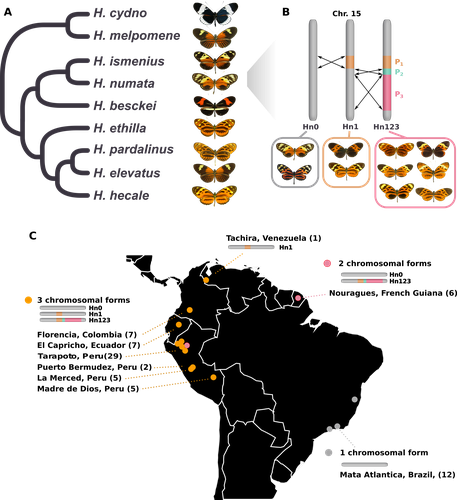
Balancing selection at a wing pattern locus is associated with major shifts in genome-wide patterns of diversity and gene flow in a butterflyIs genetic diversity enhanced by a supergene?Recommended by Chris Jiggins based on reviews by Christelle Fraïsse and 2 anonymous reviewersThe butterfly species Heliconius numata has a remarkable wing pattern polymorphism, with multiple pattern morphs all controlled by a single genetic locus, which harbours multiple inversions. Each morph is a near-perfect mimic of a species in the fairly distantly related genus of butterflies, Melinaea. The article by Rodríguez de Cara et al (2023) argues that the balanced polymorphism at this single wing patterning locus actually has a major effect on genetic diversity across the whole genome. First, polymorphic populations within H. numata are more dioverse than those without polymorphism. Second, H. numata is more genetically diverse than other related species and finally reconstruction of historical demography suggests that there has been a recent increase in effective population size, putatively associated with the acquisition of the supergene polymorphism. The supergene itself generates disassortative mating, such that morphs prefer to mate with others dissimilar to themselves - in this way it is similar to mechanisms for preventing inbreeding such as self-incompatibility loci in plants. This provides a potential mechanism whereby non-random mating patterns could increase effective population size. The authors also explore this mechanism using forward simulations, and show that mating patterns at a single locus can influence linked genetic diversity over a large scale. Overall, this is an intriguing study, which suggests a far more widespread genetic impact of a single locus than might be expected. There are interesting parallels with mechanisms of inbreeding prevention in plants, such as the Pin/Thrum polymorphism in Primula, which also rely on mating patterns determined by a single locus but presumably also influence genetic diversity genome-wide by promoting outbreeding. REFERENCES Rodríguez de Cara MÁ, Jay P, Rougemont Q, Chouteau M, Whibley A, Huber B, Piron-Prunier F, Ramos RR, Freitas AVL, Salazar C, Silva-Brandão KL, Torres TT, Joron M (2023) Balancing selection at a wing pattern locus is associated with major shifts in genome-wide patterns of diversity and gene flow. bioRxiv, 2021.09.29.462348, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.09.29.462348 | Balancing selection at a wing pattern locus is associated with major shifts in genome-wide patterns of diversity and gene flow in a butterfly | María Ángeles Rodríguez de Cara, Paul Jay, Quentin Rougemont, Mathieu Chouteau, Annabel Whibley, Barbara Huber, Florence Piron-Prunier, Renato Rogner Ramos, André V. L. Freitas, Camilo Salazar, Karina Lucas Silva-Brandão, Tatiana Texeira Torres, M... | <p style="text-align: justify;">Selection shapes genetic diversity around target mutations, yet little is known about how selection on specific loci affects the genetic trajectories of populations, including their genomewide patterns of diversity ... | | Evolutionary Ecology, Genome Evolution, Hybridization / Introgression, Population Genetics / Genomics | Chris Jiggins | 2021-10-13 17:54:33 |




