
Latest recommendations

| Id | Title | Authors▼ | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
05 Oct 2022
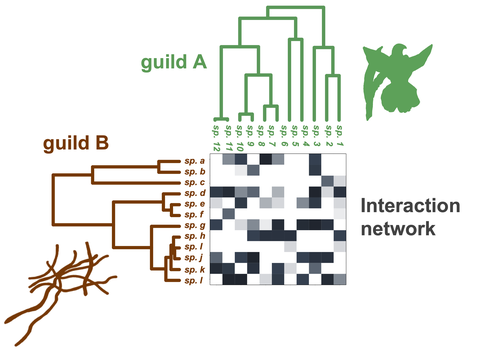
Do closely related species interact with similar partners? Testing for phylogenetic signal in bipartite interaction networksTesting for phylogenetic signal in species interaction networksRecommended by Alejandro Gonzalez Voyer based on reviews by Joaquin Calatayud and Thomas GuillermeSpecies are immersed within communities in which they interact mutualistically, as in pollination or seed dispersal, or nonreciprocally, such as in predation or parasitism, with other species and these interactions play a paramount role in shaping biodiversity (Bascompte and Jordano 2013). Researchers have become increasingly interested in the processes that shape these interactions and how these influence community structure and responses to disturbances. Species interactions are often described using bipartite interaction networks and one important question is how the evolutionary history of the species involved influences the network, including whether there is phylogenetic signal in interactions, in other words whether closely related species interact with other closely related species (Bascompte and Jordano 2013, Perez-Lamarque et al. 2022). To address this question different approaches, correlative and model-based, have been developed to test for phylogenetic signal in interactions, although comparative analyses of the performance of these different metrics are lacking. In their article Perez-Lamarque et al. (2022) set out to test the statistical performance of two widely-used methods, Mantel tests and Phylogenetic Bipartite Linear Models (PBLM; Ives and Godfray 2006) using simulations. Phylogenetic signal is measured as the degree to which distance to the nearest common ancestor predicts the observed similarity in trait values among species. In species interaction networks, the data are actually the between-species dissimilarity among interacting species (Perez-Lamarque et al. 2022), and typical approaches to test for phylogenetic signal cannot be used. However, the Mantel test provides a useful means of analyzing the correlation between two distance matrices, the between-species phylogenetic distance and the between-species dissimilarity in interactions. The PBLM approach, on the other hand, assumes that interactions between species are influenced by unobserved traits that evolve along the phylogenies following a given phenotypic evolution model and the parameters of this model are interpreted in terms of phylogenetic signal (Ives and Godfray 2006). Perez-Lamarque et al (2022) found that the model-based PBLM approach has a high type-I error rate, in other words it often detected phylogenetic signal when there was none. The simple Mantel test was found to present a low type-I error rate and moderate statistical power. However, it tended to overestimate the degree to which species interact with dissimilar partners. In addition to the aforementioned analyses, the authors also tested whether the simple Mantel test was able to detect phylogenetic signal in interactions among species within a given clade in the phylogeny, as phylogenetic signal in species interactions may be localized within specific clades. The article concludes with general guidelines for users wishing to test phylogenetic signal in their interaction networks and illustrates them with an example of an orchid-mycorrhizal fungus network from the oceanic island of La Réunion (Martos et al 2012). This broadly accessible article provides a valuable analysis of the performance of tests of phylogenetic signal in interaction networks enabling users to make informed choices of the analytical methods they wish to employ, and provide useful and detailed guidelines. Therefore, the work should be of broad interest to researchers studying species interactions. References Bascompte J, Jordano P (2013) Mutualistic Networks. Princeton University Press. https://doi.org/10.1515/9781400848720 Ives AR, Godfray HCJ (2006) Phylogenetic Analysis of Trophic Associations. The American Naturalist, 168, E1–E14. https://doi.org/10.1086/505157 Martos F, Munoz F, Pailler T, Kottke I, Gonneau C, Selosse M-A (2012) The role of epiphytism in architecture and evolutionary constraint within mycorrhizal networks of tropical orchids. Molecular Ecology, 21, 5098–5109. https://doi.org/10.1111/j.1365-294X.2012.05692.x Perez-Lamarque B, Maliet O, Pichon B, Selosse M-A, Martos F, Morlon H (2022) Do closely related species interact with similar partners? Testing for phylogenetic signal in bipartite interaction networks. bioRxiv, 2021.08.30.458192, ver. 6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.08.30.458192 | Do closely related species interact with similar partners? Testing for phylogenetic signal in bipartite interaction networks | Benoît Perez-Lamarque, Odile Maliet, Benoît Pichon, Marc-André Selosse, Florent Martos, Hélène Morlon | <p style="text-align: justify;">Whether interactions between species are conserved on evolutionary time-scales has spurred the development of both correlative and process-based approaches for testing phylogenetic signal in interspecific interactio... | | Evolutionary Ecology, Species interactions | Alejandro Gonzalez Voyer | 2022-03-10 13:48:15 | ||
28 Feb 2023
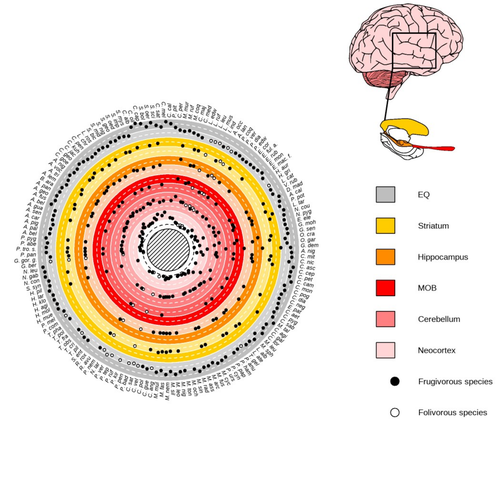
Primate sympatry shapes the evolution of their brain architectureMacroevolutionary drivers of brain evolution in primatesRecommended by Fabien Condamine based on reviews by Paula Gonzalez, Orlin Todorov and 3 anonymous reviewersStudying the evolution of animal cognition is challenging because many environmental and species-related factors can be intertwined, which is further complicated when looking at deep-time evolution. Previous knowledge has emphasized the role of intraspecific interactions in affecting the socio-ecological environment shaping cognition. However, much less is known about such an effect at the interspecific level. Yet, the coexistence of different species in the same geographic area at a given time (sympatry) can impact the evolutionary history of species through character displacement due to biotic interactions. Trait evolution has been observed and tested with morphological external traits but more rarely with brain evolution. Compared to most species’ traits, brain evolution is even more delicate to assess since specific brain regions can be involved in different functions, may they be individual-based and social-based information processing. In a very original and thoroughly executed study, Robira & Perez-Lamarque (2023) addressed the question: How does the co-occurrence of congeneric species shape brain evolution and influence species diversification? By considering brain size as a proxy for cognition, they evaluated whether species sympatry impacted the evolution of cognition in frugivorous primates. Fruit resources are hard to find, not continuous through time, heterogeneously distributed across space, but can be predictable. Hence, cognition considerably shapes the foraging strategy and competition for food access can be fierce. Over long timescales, it remains unclear whether brain size and the pace of species diversification are linked in the context of sympatry, and if so how. Recent studies have found that larger brain sizes can be associated with higher diversification rates in birds (Sayol et al. 2019). Similarly, Robira & Perez-Lamarque (2023) thus wondered if the evolution of brain size in primates impacted their dynamic of species diversification, which has been suggested (Melchionna et al. 2020) but not tested. Prior to anything, Robira & Perez-Lamarque (2023) had to retrace the evolutionary history of sympatry between frugivorous primate lineages through time using the primate tree of life, species’ extant distribution, and process-based models to estimate ancestral range evolution. To infer the effect of species sympatry on the evolution of cognition in frugivorous primates, the authors evaluated the support for phylogenetic models of brain size evolution accounting or not for species sympatry and investigated the directionality of the selection induced by sympatry on brain size evolution. Finally, to better understand the impact of cognition and interactions between primates on their evolutionary success, they tested for correlations between brain size or species’ sympatry and species diversification. Robira & Perez-Lamarque (2023) found that the evolution of the whole brain or brain regions used in immediate information processing was best fitted with models not considering sympatry. By contrast, models considering species sympatry best predicted the evolution of brain regions related to long-term memory of interactions with the socio-ecological environment, with a decrease in their size along with stronger sympatry. Specifically, they found that sympatry was associated with a decrease in the relative size of the hippocampus and striatum, but had no significant effect on the neocortex, cerebellum, or overall brain size. The hippocampus is a brain region that plays a crucial role in processing and memorizing spatiotemporal information, which is relevant for frugivorous primates in their foraging behavior. The study suggests that competition between sympatric species for limited food resources may lead to a more complex and unpredictable food distribution, which may in turn render cognitive foraging not advantageous and result in a selection for smaller brain regions involved in foraging. Niche partitioning and dietary specialization in sympatry may also impact cognitive abilities, with more specialized diets requiring lower cognitive abilities and smaller brain region sizes. On the other hand, the absence of an effect of sympatry on brain regions involved in immediate sensory information processing, such as the cerebellum and neocortex, suggests that foragers do not exploit cues left out by sympatric heterospecific species, or they may discard environmental cues in favor of social cues. This is a remarkable study that highlights the importance of considering the impact of ecological factors, such as sympatry, on the evolution of specific brain regions involved in cognitive processes, and the potential trade-offs in brain region size due to niche partitioning and dietary specialization in sympatry. Further research is needed to explore the mechanisms behind these effects and to test for the possible role of social cues in the evolution of brain regions. This study provides insights into the selective pressures that shape brain evolution in primates. References Melchionna M, Mondanaro A, Serio C, Castiglione S, Di Febbraro M, Rook L, Diniz-Filho JAF, Manzi G, Profico A, Sansalone G, Raia P (2020) Macroevolutionary trends of brain mass in Primates. Biological Journal of the Linnean Society, 129, 14–25. https://doi.org/10.1093/biolinnean/blz161 Robira B, Perez-Lamarque B (2023) Primate sympatry shapes the evolution of their brain architecture. bioRxiv, 2022.05.09.490912, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.05.09.490912 Sayol F, Lapiedra O, Ducatez S, Sol D (2019) Larger brains spur species diversification in birds. Evolution, 73, 2085–2093. https://doi.org/10.1111/evo.13811 | Primate sympatry shapes the evolution of their brain architecture | Benjamin Robira, Benoit Perez-Lamarque | <p style="text-align: justify;">The main hypotheses on the evolution of animal cognition emphasise the role of conspecifics in affecting the socio-ecological environment shaping cognition. Yet, space is often simultaneously occupied by multiple sp... | | Behavior & Social Evolution, Bioinformatics & Computational Biology, Evolutionary Ecology, Macroevolution, Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Fabien Condamine | 2022-05-10 13:43:02 | ||
16 Dec 2016
POSTPRINT
Spatiotemporal microbial evolution on antibiotic landscapesA poster child for experimental evolutionRecommended by Daniel Rozen and Arjan de VisserEvolution is usually studied via two distinct approaches: by inferring evolutionary processes from relatedness patterns among living species or by observing evolution in action in the laboratory or field. A recent study by Baym and colleagues in Science [1] has now combined these approaches by taking advantage of the pattern left behind by spatially evolving bacterial populations. Evolution is often considered too slow to see, and can only be inferred by studying patterns of relatedness using phylogenetic trees. Increasingly, however, researchers are moving nature into the lab and watching as evolution unfolds under their noses. The field of experimental evolution follows evolutionary change in the laboratory over 10s to 1000s of generations, yielding insights into bacterial, viral, plant, or fly evolution (among many other species) that are simply not possible in the field. Yet, as powerful as experimental evolution is, it lacks a posterchild. There is no Galapagos finch radiation, nor a stunning series of cichlids to showcase to our students to pique their interests. Let’s face it, E. coli is no stickleback! And while practitioners of experimental evolution can explain the virtues of examining 60,000 generations of bacterial evolution in action, appreciating this nevertheless requires a level of insight and imagination that often eludes students, who need to see “it” to get it. Enter MEGA, an idea and a film that could become the new face of experimental evolution. It replaces big numbers of generations or images of scientists, with an actual picture of the scientific result. MEGA, or Microbial Evolution and Growth Arena, is essentially an enormous petri dish and is the brainchild of Michael Baym, Tami Leiberman and their colleagues in Roy Kishony’s lab at Technion Israel Institute of Technology and Harvard Medical School. The idea of MEGA is to allow bacteria to swim over a spatially defined landscape while adapting to the local conditions, in this case antibiotics. When bacteria are inoculated onto one end of the plate they consume resources while swarming forward from the plate edge. In a few days, the bacteria grow into an area with antibiotics to which they are susceptible. This stops growth until a mutation arises that permits the bacteria to jump this hurdle, after which growth proceeds until the next hurdle of a 10-fold higher antibiotic concentration, and so on. By this simple approach, Baym et al. [1] evolved E. coli that were nearly 105-fold more resistant to two different antibiotics in just over 10 days. In addition, they identified the mutations that were required for these changes, showed that mutations conferring smaller benefits were required before bacteria could evolve maximal resistance, observed changes to the mutation rate, and demonstrated the importance of spatial structure in constraining adaptation. For one thing, the rate of resistance evolution is impressive, and also quite scary given the mounting threat of antibiotic-resistant pathogens. However, MEGA also offers a uniquely visual insight into evolutionary change. By taking successive images of the MEGA plate, the group was able to watch the bacteria move, get trapped because of their susceptibility to the antibiotic, and then get past these traps as new mutations emerged that increased resistance. Each transition showcases evolution in real time. In addition, by leaving a spatial pattern of evolutionary steps behind, the MEGA plate offers unique opportunities to thoroughly investigate these steps when the experiment is finished. For instance, subsequent steps in mutational pathways can be characterized, but also their effects on fitness can be quantified in situ by measuring changes in survival and reproduction. This new method is undoubtedly a boon to the field of experimental evolution and offers endless opportunities for experimental elaboration. Perhaps of equal importance, MEGA is a tool that is portable to the classroom and to the public at large. Don’t believe in evolution? Watch this. You only have time for a short internship or lab practical? No problem. Don’t worry much about antibiotic resistance? Check this out. Like the best experimental tools, MEGA is simple but allows for complicated insights. And even if it is less charismatic than a finch, it still allows for the kinds of “gee-whiz” insights that will get students hooked on evolutionary biology. Reference [1] Baym M, Lieberman TD, Kelsic ED, Chait R, Gross R, Yelin I, Kishony R. 2016. Spatiotemporal microbial evolution on antibiotic landscapes. Science 353:1147-1151. doi: 10.1126/science.aag0822 | Spatiotemporal microbial evolution on antibiotic landscapes | Baym M, Lieberman TD, Kelsic ED, Chait R, Gross R, Yelin I, Kishony R | A key aspect of bacterial survival is the ability to evolve while migrating across spatially varying environmental challenges. Laboratory experiments, however, often study evolution in well-mixed systems. Here, we introduce an experimental device,... | | Adaptation, Evolutionary Applications, Experimental Evolution | Daniel Rozen | 2016-12-14 14:26:06 | ||
24 Mar 2023
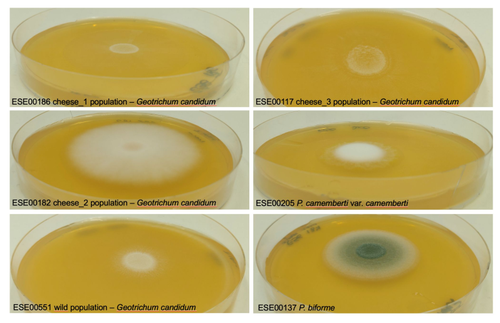
Domestication of different varieties in the cheese-making fungus Geotrichum candidumDiverse outcomes in cheese fungi domesticationRecommended by Christelle Fraïsse based on reviews by Delphine Sicard and 1 anonymous reviewer based on reviews by Delphine Sicard and 1 anonymous reviewer
Domestication is a complex process that imprints the demography and the genomes of domesticated populations, enforcing strong selective pressures on traits favourable to humans, e.g. for food production [1]. Domestication has been quite intensely studied in plants and animals, but less so in micro-organisms such as fungi, despite their assets (e.g. their small genomes and tractability in the lab). This elegant study by Bennetot and collaborators [2] on the cheese-making fungus Geotrichum candidum adds to the mounting body of studies in the genomics of fungi, proving they are excellent models in evolutionary biology for studying adaptation and drift in eukaryotes [3]. Bennetot et al. newly showed with whole genome sequences that all G. candidum strains isolated from cheese form a monophyletic clade subdivided into three genetically differentiated populations with several admixed strains, while the wild strains sampled from diverse geographic locations form a sister clade. This suggests the wild progenitor was not sampled in the present study and calls for future exciting work on the domestication history of the G. candidum fungus. The authors scanned the genomes for footprints of adaptation to the cheese environment and identified promising candidates, such as a gene involved in iron uptake (this element is limiting in cheese). Their functional genome analysis also provides evidence for higher contents of transposable elements in cheese-making strains, likely due to relaxed selection during the domestication process. This paper is particularly impressive in that the authors complemented the population genomic approach with the phenotypic characterization of the strains and tested their ability to outcompete common fungal food spoilers. The authors convincingly showed that cheese-making strains display phenotypic differences relative to wild relatives for multiple traits such as slower growth, lower proteolysis activity and a greater amount of volatiles attractive to consumers, these phenotypes being beneficial for cheese making. Finally, this work is particularly inspiring because it thoroughly discusses convergent evolution during domestication in different cheese-associated fungi. Indeed, studying populations experiencing similar environmental pressures is fundamental to understanding whether evolution is repeatable [4]. For instance, all three cheese populations of G. candidum exhibit a lower genetic diversity than wild populations. However, only one population displays a stronger domestication syndrome, resembling the Penicillium camemberti situation [5]. Furthermore, different cheese-making practices may have led to varying situations with clonal lineages in non-Roquefort P. roqueforti and P. camemberti [5, 6], while the cheese-making G. candidum populations still harbour some diversity. In a nutshell, Bennetot's study makes an important contribution to evolutionary biology and highlights the value of diversifying our model organisms toward under-represented clades. REFERENCES [1] Diamond J (2002) Evolution, consequences and future of plant and animal domestication. Nature 418: 700–707. https://doi.org/10.1038/nature01019 [2] Bennetot B, Vernadet J-P, Perkins V, Hautefeuille S, Rodríguez de la Vega RC, O’Donnell S, Snirc A, Grondin C, Lessard M-H, Peron A-C, Labrie S, Landaud S, Giraud T, Ropars J (2023) Domestication of different varieties in the cheese-making fungus Geotrichum candidum. bioRxiv, 2022.05.17.492043, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.05.17.492043 [3] Gladieux P, Ropars J, Badouin H, Branca A, Aguileta G, de Vienne DM, Rodríguez de la Vega RC, Branco S, Giraud T (2014) Fungal evolutionary genomics provides insight into the mechanisms of adaptive divergence in eukaryotes. Mol. Ecol. 23: 753–773. https://doi.org/10.1111/mec.12631 [4] Bolnick DI, Barrett RD, Oke KB, Rennison DJ, Stuart YE (2018) (Non)Parallel evolution. Ann. Rev. Ecol. Evol. Syst. 49: 303–330. https://doi.org/10.1146/annurev-ecolsys-110617-062240 [5] Ropars J, Didiot E, Rodríguez de la Vega RC, Bennetot B, Coton M, Poirier E, Coton E, Snirc A, Le Prieur S, Giraud T (2020) Domestication of the Emblematic White Cheese-Making Fungus Penicillium camemberti and Its Diversification into Two Varieties. Current Biol. 30: 4441–4453.e4. https://doi.org/10.1016/j.cub.2020.08.082 [6] Dumas, E, Feurtey, A, Rodríguez de la Vega, RC, Le Prieur S, Snirc A, Coton M, Thierry A, Coton E, Le Piver M, Roueyre D, Ropars J, Branca A, Giraud T (2020) Independent domestication events in the blue-cheese fungus Penicillium roqueforti. Mol Ecol. 29: 2639–2660. https://doi.org/10.1111/mec.15359 | Domestication of different varieties in the cheese-making fungus *Geotrichum candidum* | Bastien Bennetot, Jean-Philippe Vernadet, Vincent Perkins, Sophie Hautefeuille, Ricardo C. Rodríguez de la Vega, Samuel O’Donnell, Alodie Snirc, Cécile Grondin, Marie-Hélène Lessard, Anne-Claire Peron, Steve Labrie, Sophie Landaud, Tatiana Giraud,... | <p>Domestication is an excellent model for studying adaptation processes, involving recent adaptation and diversification, convergence following adaptation to similar conditions, as well as degeneration of unused functions. <em>Geotrichum candidum... | | Adaptation, Genome Evolution, Population Genetics / Genomics | Christelle Fraïsse | 2022-08-12 20:50:42 | ||
05 Feb 2021
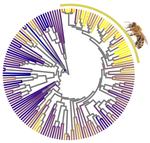
Relaxation of purifying selection suggests low effective population size in eusocial Hymenoptera and solitary pollinating beesMulti-gene and lineage comparative assessment of the strength of selection in HymenopteraRecommended by Bertanne Visser based on reviews by Michael Lattorff and 1 anonymous reviewerGenetic variation is the raw material for selection to act upon and the amount of genetic variation present within a population is a pivotal determinant of a population’s evolutionary potential. A large effective population size, i.e., the ideal number of individuals experiencing the same amount of genetic drift and inbreeding as an actual population, Ne (Wright 1931, Crow 1954), thus increases the probability of long-term survival of a population. However, natural populations, as opposed to theoretical ones, rarely adhere to the requirements of an ideal panmictic population (Sjödin et al. 2005). A range of circumstances can reduce Ne, including the structuring of populations (through space and time, as well as age and developmental stages) and inbreeding (Charlesworth 2009). In mammals, species with a larger body mass (as a proxy for lower Ne) were found to have a higher rate of nonsynonymous nucleotide substitutions (that alter the amino acid sequence of a protein), as well as radical amino acid substitutions (altering the physicochemical properties of a protein) (Popadin et al. 2007). In general, low effective population sizes increase the chance of mutation accumulation and drift, while reducing the strength of selection (Sjödin et al. 2005). References Charlesworth, B. (2009). Effective population size and patterns of molecular evolution and variation. Nature Reviews Genetics, 10(3), 195-205. doi: https://doi.org/10.1038/nrg2526 | Relaxation of purifying selection suggests low effective population size in eusocial Hymenoptera and solitary pollinating bees | Arthur Weyna, Jonathan Romiguier | <p>With one of the highest number of parasitic, eusocial and pollinator species among all insect orders, Hymenoptera features a great diversity of lifestyles. At the population genetic level, such life-history strategies are expected to decrease e... | | Behavior & Social Evolution, Genome Evolution, Life History, Molecular Evolution, Population Genetics / Genomics | Bertanne Visser | 2020-04-21 17:30:57 | ||
31 Mar 2022
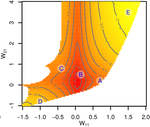
Gene network robustness as a multivariate characterGenetic and environmental robustness are distinct yet correlated evolvable traits in a gene networkRecommended by Frédéric Guillaume based on reviews by Diogo Melo, Charles Mullon and Charles Rocabert
Organisms often show robustness to genetic or environmental perturbations. Whether these two components of robustness can evolve separately is the focus of the paper by Le Rouzic [1]. Using theoretical analysis and individual-based computer simulations of a gene regulatory network model, he shows that multiple aspects of robustness can be investigated as a set of pleiotropically linked quantitative traits. While genetically correlated, various robustness components (e.g., mutational, developmental, homeostasis) of gene expression in the regulatory network evolved more or less independently from each other under directional selection. The quantitative approach of Le Rouzic could explain both how unselected robustness components can respond to selection on other components and why various robustness-related features seem to have their own evolutionary history. Moreover, he shows that all components were evolvable, but not all to the same extent. Robustness to environmental disturbances and gene expression stability showed the largest responses while increased robustness to genetic disturbances was slower. Interestingly, all components were positively correlated and remained so after selection for increased or decreased robustness. This study is an important contribution to the discussion of the evolution of robustness in biological systems. While it has long been recognized that organisms possess the ability to buffer genetic and environmental perturbations to maintain homeostasis (e.g., canalization [2]), the genetic basis and evolutionary routes to robustness and canalization are still not well understood. Models of regulatory gene networks have often been used to address aspects of robustness evolution (e.g., [3]). Le Rouzic [1] used a gene regulatory network model derived from Wagner’s model [4]. The model has as end product the expression level of a set of genes influenced by a set of regulatory elements (e.g., transcription factors). The level and stability of expression are a property of the regulatory interactions in the network. Le Rouzic made an important contribution to the study of such gene regulation models by using a quantitative genetics approach to the evolution of robustness. He crafted a way to assess the mutational variability and selection response of the components of robustness he was interested in. Le Rouzic’s approach opens avenues to investigate further aspects of gene network evolutionary properties, for instance to understand the evolution of phenotypic plasticity. Le Rouzic also discusses ways to measure his different robustness components in empirical studies. As the model is about gene expression levels at a set of protein-coding genes influenced by a set of regulatory elements, it naturally points to the possibility of using RNA sequencing to measure the variation of gene expression in know gene networks and assess their robustness. Robustness could then be studied as a multidimensional quantitative trait in an experimental setting. References [1] Le Rouzic, A (2022) Gene network robustness as a multivariate character. arXiv: 2101.01564, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://arxiv.org/abs/2101.01564 [2] Waddington CH (1942) Canalization of Development and the Inheritance of Acquired Characters. Nature, 150, 563–565. https://doi.org/10.1038/150563a0 [3] Draghi J, Whitlock M (2015) Robustness to noise in gene expression evolves despite epistatic constraints in a model of gene networks. Evolution, 69, 2345–2358. https://doi.org/10.1111/evo.12732 [4] Wagner A (1994) Evolution of gene networks by gene duplications: a mathematical model and its implications on genome organization. Proceedings of the National Academy of Sciences, 91, 4387–4391. https://doi.org/10.1073/pnas.91.10.4387 | Gene network robustness as a multivariate character | Arnaud Le Rouzic | <p style="text-align: justify;">Robustness to genetic or environmental disturbances is often considered as a key property of living systems. Yet, in spite of being discussed since the 1950s, how robustness emerges from the complexity of genetic ar... | | Bioinformatics & Computational Biology, Evolutionary Theory, Genotype-Phenotype, Quantitative Genetics | Frédéric Guillaume | Charles Mullon, Charles Rocabert, Diogo Melo | 2021-01-11 17:48:20 | |
07 Nov 2019

New insights into the population genetics of partially clonal organisms: when seagrass data meet theoretical expectationsInferring rates of clonal versus sexual reproduction from population genetics dataRecommended by Olivier J Hardy based on reviews by Ludwig TRIEST, Stacy Krueger-Hadfield and 1 anonymous reviewerIn partially clonal organisms, genetic markers are often used to characterize the genotypic diversity of populations and infer thereof the relative importance of clonal versus sexual reproduction. Most studies report a measure of genotypic diversity based on a ratio, R, of the number of distinct multilocus genotypes over the sample size, and qualitatively interpret high / low R as indicating the prevalence of sexual / clonal reproduction. However, a theoretical framework allowing to quantify the relative rates of clonal versus sexual reproduction from genotypic diversity is still lacking, except using temporal sampling. Moreover, R is intrinsically highly dependent on sample size and sample design, while alternative measures of genotypic diversity are more robust to sample size, like D*, which is equivalent to the Gini-Simpson diversity index applied to multilocus genotypes. Another potential indicator of reproductive strategies is the inbreeding coefficient, Fis, because population genetics theory predicts that clonal reproduction should lead to negative Fis, at least when the sexual reproduction component occurs through random mating. Taking advantage of this prediction, Arnaud-Haond et al. [1] reanalysed genetic data from 165 populations of four partially clonal seagrass species sampled in a standardized way. They found positive correlations between Fis and both R and D* within each species, reflecting variation in the relative rates of sexual versus clonal reproduction among populations. Moreover, the differences of mean genotypic diversity and Fis values among species were also consistent with their known differences in reproductive strategies. Arnaud-Haond et al. [1] also conclude that previous works based on the interpretation of R generally lead to underestimate the prevalence of clonality in seagrasses. Arnaud-Haond et al. [1] confirm experimentally that Fis merits to be interpreted more properly than usually done when inferring rates of clonal reproduction from population genetics data of species reproducing both sexually and clonally. An advantage of Fis is that it is much less affected by sample size than R, and thus should be more reliable when comparing studies differing in sample design. Hence, when the rate of clonal reproduction becomes significant, we expect Fis < 0 and D* < 1. I expect these two indicators of clonality to be complementary because they rely on different consequences of clonality on pattern of genetic variation. Nevertheless, both measures can be affected by other factors. For example, null alleles, selfing or biparental inbreeding can pull Fis upwards, potentially eliminating the signature of clonal reproduction. Similarly, D* (and other measures of genotypic diversity) can be low because the polymorphism of the genetic markers used is too limited or because sexual reproduction often occurs through selfing, eventually resulting in highly similar homozygous genotypes. References [1] Arnaud-Haond, S., Stoeckel, S., and Bailleul, D. (2019). New insights into the population genetics of partially clonal organisms: when seagrass data meet theoretical expectations. ArXiv:1902.10240 [q-Bio], v6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. Retrieved from http://arxiv.org/abs/1902.10240 | New insights into the population genetics of partially clonal organisms: when seagrass data meet theoretical expectations | Arnaud-Haond, Sophie, Stoeckel, Solenn, and Bailleul, Diane | <p>Seagrass meadows are among the most important coastal ecosystems, in terms of both spatial extent and ecosystem services, but they are also declining worldwide. Understanding the drivers of seagrass meadow dynamics is essential for designing so... | | Evolutionary Ecology, Population Genetics / Genomics, Reproduction and Sex | Olivier J Hardy | 2019-03-01 21:57:34 | ||
06 Jun 2019
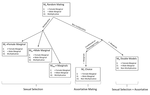
Multi-model inference of non-random mating from an information theoretic approachTell me who you mate with, I’ll tell you what’s going onRecommended by Sara Magalhaes and Alexandre Courtiol based on reviews by Alexandre Courtiol and 2 anonymous reviewersThe study of sexual selection goes as far as Darwin himself. Since then, elaborate theories concerning both intra- and inter-sexual sexual have been developed, and elegant experiments have been designed to test this body of theory. It may thus come as a surprise that the community is still debating on the correct way to measure simple components of sexual selection, such as the Bateman gradient (i.e., the covariance between the number of matings and the number of offspring)[1,2], or to quantify complex behaviours such as mate choice (the non-random choice of individuals with particular characters as mates)[3,4] and their consequences. References [1] Bateman, A. J. (1948). Intra-sexual selection in Drosophila. Heredity, 2(3), 349-368. doi: 10.1038/hdy.1948.21 | Multi-model inference of non-random mating from an information theoretic approach | Antonio Carvajal-Rodríguez | <p>Non-random mating has a significant impact on the evolution of organisms. Here, I developed a modelling framework for discrete traits (with any number of phenotypes) to explore different models connecting the non-random mating causes (mate comp... | | Evolutionary Ecology, Evolutionary Theory, Sexual Selection | Sara Magalhaes | 2019-02-08 19:24:03 | ||
08 Feb 2019
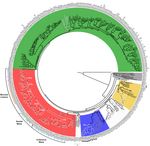
Genome plasticity in Papillomaviruses and de novo emergence of E5 oncogenesE5, the third oncogene of PapillomavirusRecommended by Hirohisa Kishino based on reviews by Leonardo de Oliveira Martins and 1 anonymous reviewerPapillomaviruses (PVs) infect almost all mammals and possibly amniotes and bony fishes. While most of them have no significant effects on the hosts, some induce physical lesions. Phylogeny of PVs consists of a few crown groups [1], among which AlphaPVs that infect primates including human have been well studied. They are associated to largely different clinical manifestations: non-oncogenic PVs causing anogenital warts, oncogenic and non-oncogenic PVs causing mucosal lesions, and non-oncogenic PVs causing cutaneous warts. References [1] Bravo, I. G., & Alonso, Á. (2004). Mucosal human papillomaviruses encode four different E5 proteins whose chemistry and phylogeny correlate with malignant or benign growth. Journal of virology, 78, 13613-13626. doi: 10.1128/JVI.78.24.13613-13626.2004 | Genome plasticity in Papillomaviruses and de novo emergence of E5 oncogenes | Anouk Willemsen, Marta Félez-Sánchez, and Ignacio G. Bravo | <p>The clinical presentations of papillomavirus (PV) infections come in many different flavors. While most PVs are part of a healthy skin microbiota and are not associated to physical lesions, other PVs cause benign lesions, and only a handful of ... | | Genome Evolution, Molecular Evolution, Phylogenetics / Phylogenomics | Hirohisa Kishino | 2018-06-04 16:15:39 | ||
03 Oct 2018
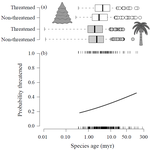
Range size dynamics can explain why evolutionarily age and diversification rate correlate with contemporary extinction risk in plantsAre both very young and the very old plant lineages at heightened risk of extinction?Recommended by Arne Mooers based on reviews by Dan Greenberg and 1 anonymous reviewerHuman economic activity is responsible for the vast majority of ongoing extinction, but that does not mean lineages are being affected willy-nilly. For amphibians [1] and South African flowering plants [2], young species have a somewhat higher than expected chance of being threatened with extinction. In contrast, older Australian marsupial lineages seem to be more at risk [3]. Both of the former studies suggested that situations leading to peripheral isolation might simultaneously increase ongoing speciation and current threat via small geographic range, while the authors of the latter study suggested that older species might have evolved increasingly narrow niches. Here, Andrew Tanentzap and colleagues [4] dig deeper into the putative links between species age, niche breadth and threat status. Across 500-some plant genera worldwide, they find that, indeed, ""younger"" species (i.e. from younger and faster-diversifying genera) were more likely to be listed as imperiled by the IUCN, consistent with patterns for amphibians and African plants. Given this, results from their finer-level analyses of conifers are initially bemusing: here, ""older"" (i.e., on longer terminal branches) species were at higher risk. This would make conifers more like Australian marsupials, with the rest of the plants being more like amphibians. However, here where the data were more finely grained, the authors detected a second interesting pattern: using an intriguing matched-pair design, they detect a signal of conifer species niches seemingly shrinking as a function of age. The authors interpret this as consistent with increasing specialization, or loss of ancestral warm wet habitat, over paleontological time. It is true that conifers in general are older than plants more generally, with some species on branches that extend back many 10s of millions of years, and so a general loss of suitable habitat makes some sense. If so, both the pattern for all plants (small initial ranges heightening extinction) and the pattern for conifers (eventual increasing specialization or habitat contraction heightening extinction) could occur, each on a different time scale. As a coda, the authors detected no effect of age on threat status in palms; however, this may be both because palms have already lost species to climate-change induced extinction, and because they are thought to speciate more via long-distance dispersal and adaptive divergence then via peripheral isolation. References [1] Greenberg, D. A., & Mooers, A. Ø. (2017). Linking speciation to extinction: Diversification raises contemporary extinction risk in amphibians. Evolution Letters, 1, 40–48. doi: 10.1002/evl3.4 | Range size dynamics can explain why evolutionarily age and diversification rate correlate with contemporary extinction risk in plants | Andrew J. Tanentzap, Javier Igea, Matthew G. Johnston, Matthew J. Larcombe | <p>Extinction threatens many species, yet few factors predict this risk across the plant Tree of Life (ToL). Taxon age is one factor that may associate with extinction if occupancy of geographic and adaptive zones varies with time, but evidence fo... | | Macroevolution, Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Arne Mooers | 2018-02-01 21:01:19 |




