
Latest recommendations
| Id | Title * | Authors * | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
18 Dec 2017
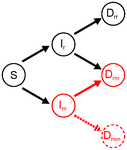
Co-evolution of virulence and immunosuppression in multiple infectionsTwo parasites, virulence and immunosuppression: how does the whole thing evolve?Recommended by Sara Magalhaes based on reviews by 2 anonymous reviewersHow parasite virulence evolves is arguably the most important question in both the applied and fundamental study of host-parasite interactions. Typically, this research area has been progressing through the formalization of the problem via mathematical modelling. This is because the question is a complex one, as virulence is both affected and affects several aspects of the host-parasite interaction. Moreover, the evolution of virulence is a problem in which ecology (epidemiology) and evolution (changes in trait values through time) are tightly intertwined, generating what is now known as eco-evolutionary dynamics. Therefore, intuition is not sufficient to address how virulence may evolve. References [1] Anderson RM and May RM. 1982. Coevolution of hosts and parasites. Parasitology, 1982. 85: 411–426. doi: 10.1017/S0031182000055360 [2] Kamiya T, Mideo N and Alizon S. 2017. Coevolution of virulence and immunosuppression in multiple infections. bioRxiv, ver. 7 peer-reviewed by PCI Evol Biol, 149211. doi: 10.1101/139147 | Co-evolution of virulence and immunosuppression in multiple infections | Tsukushi Kamiya, Nicole Mideo, Samuel Alizon | Many components of the host-parasite interaction have been shown to affect the way virulence, that is parasite induced harm to the host, evolves. However, co-evolution of multiple traits is often neglected. We explore how an immunosuppressive mech... | | Evolutionary Applications, Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Epidemiology, Evolutionary Theory | Sara Magalhaes | 2017-06-13 16:49:45 | ||
05 Dec 2017

Reconstruction of body mass evolution in the Cetartiodactyla and mammals using phylogenomic dataPredicting small ancestors using contemporary genomes of large mammalsRecommended by Bruce Rannala based on reviews by Bruce Rannala and 1 anonymous reviewer based on reviews by Bruce Rannala and 1 anonymous reviewer
Recent methodological developments and increased genome sequencing efforts have introduced the tantalizing possibility of inferring ancestral phenotypes using DNA from contemporary species. One intriguing application of this idea is to exploit the apparent correlation between substitution rates and body size to infer ancestral species' body sizes using the inferred patterns of substitution rate variation among species lineages based on genomes of extant species [1]. References [1] Romiguier J, Ranwez V, Douzery EJP and Galtier N. 2013. Genomic evidence for large, long-lived ancestors to placental mammals. Molecular Biology and Evolution 30: 5–13. doi: 10.1093/molbev/mss211 [2] Figuet E, Ballenghien M, Lartillot N and Galtier N. 2017. Reconstruction of body mass evolution in the Cetartiodactyla and mammals using phylogenomic data. bioRxiv, ver. 3 of 4th December 2017. 139147. doi: 10.1101/139147 | Reconstruction of body mass evolution in the Cetartiodactyla and mammals using phylogenomic data | Emeric Figuet, Marion Ballenghien, Nicolas Lartillot, Nicolas Galtier | <p>Reconstructing ancestral characters on a phylogeny is an arduous task because the observed states at the tips of the tree correspond to a single realization of the underlying evolutionary process. Recently, it was proposed that ancestral traits... | | Genome Evolution, Life History, Macroevolution, Molecular Evolution, Phylogenetics / Phylogenomics | Bruce Rannala | 2017-05-18 15:28:58 | ||
20 Nov 2017
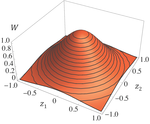
Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selectionUnderstanding genetic variance, load, and inbreeding depression with selfingRecommended by Aneil F. Agrawal based on reviews by Frédéric Guillaume and 1 anonymous reviewerA classic problem in evolutionary biology is to understand the genetic variance in fitness. The simplest hypothesis is that variation exists, even in well-adapted populations, as a result of the balance between mutational input and selective elimination. This variation causes a reduction in mean fitness, known as the mutation load. Though mutation load is difficult to quantify empirically, indirect evidence of segregating genetic variation in fitness is often readily obtained by comparing the fitness of inbred and outbred offspring, i.e., by measuring inbreeding depression. Mutation-selection balance models have been studied as a means of understanding the genetic variance in fitness, mutation load, and inbreeding depression. Since their inception, such models have increased in sophistication, allowing us to ask these questions under more realistic and varied scenarios. The new theoretical work by Abu Awad and Roze [1] is a substantial step forward in understanding how arbitrary levels of self-fertilization affect variation, load and inbreeding depression under mutation-selection balance. References [1] Abu Awad D and Roze D. 2017. Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selection. bioRxiv, 180000, ver. 4 of 17th November 2017. doi: 10.1101/180000 [2] Lande R. 1977. The influence of the mating system on the maintenance of genetic variability in polygenic characters. Genetics 86: 485–498. [3] Charlesworth D and Charlesworth B. 1987. Inbreeding depression and its evolutionary consequences. Annual Review of Ecology and Systematics. 18: 237–268. doi: 10.1111/10.1146/annurev.es.18.110187.001321 [4] Lande R and Porcher E. 2015. Maintenance of quantitative genetic variance under partial self-fertilization, with implications for the evolution of selfing. Genetics 200: 891–906. doi: 10.1534/genetics.115.176693 [5] Roze D. 2015. Effects of interference between selected loci on the mutation load, inbreeding depression, and heterosis. Genetics 201: 745–757. doi: 10.1534/genetics.115.178533 [6] Martin G and Lenormand T. 2006. A general multivariate extension of Fisher's geometrical model and the distribution of mutation fitness effects across species. Evolution 60: 893–907. doi: 10.1111/j.0014-3820.2006.tb01169.x [7] Martin G, Elena SF and Lenormand T. 2007. Distributions of epistasis in microbes fit predictions from a fitness landscape model. Nature Genetics 39: 555–560. doi: 10.1038/ng1998 | Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selection | Diala Abu Awad and Denis Roze | The mating system of a species is expected to have important effects on its genetic diversity. In this paper, we explore the effects of partial selfing on the equilibrium genetic variance Vg, mutation load L and inbreeding depression δ under stabi... | | Evolutionary Theory, Population Genetics / Genomics, Quantitative Genetics, Reproduction and Sex | Aneil F. Agrawal | 2017-08-26 09:29:20 | ||
17 Nov 2017

ABC random forests for Bayesian parameter inferenceMachine learning methods are useful for Approximate Bayesian Computation in evolution and ecologyRecommended by Michael Blum ? based on reviews by Dennis Prangle and Michael Blum ?It is my pleasure to recommend the paper by Raynal et al. [1] about using random forest for parameter inference. There are two reviews about the paper, one review written by Dennis Prangle and another review written by myself. Both reviews were positive and included comments that have been addressed in the current version of the preprint. The paper nicely shows that modern machine learning approaches are useful for Approximate Bayesian Computation (ABC) and more generally for simulation-driven parameter inference in ecology and evolution. The authors propose to consider the random forest approach, proposed by Meinshausen [2] to perform quantile regression. The numerical implementation of ABC with random forest, available in the abcrf package, is based on the RANGER R package that provides a fast implementation of random forest for high-dimensional data. According to my reading of the manuscript, there are 3 main advantages when using random forest (RF) for parameter inference with ABC. The first advantage is that RF can handle many summary statistics and that dimension reduction is not needed when using RF. The second advantage is very nicely displayed in Figure 5, which shows the main result of the paper. If correct, 95% posterior credibility intervals (C.I.) should contain 95% of the parameter values used in simulations. Figure 5 shows that posterior C.I. obtained with rejection are too large compared to other methods. By contrast, C.I. obtained with regression methods have been shrunken. However, the shrinkage can be excessive for the smallest tolerance rates, with coverage values that can be equal to 85% instead of the expected 95% value. The attractive property of RF is that C.I. have been shrunken but the coverage is of 100% resulting in a conservative decision about parameter values. The last advantage is that no hyperparameter should be chosen. It is a parameter free approach, which is desirable because of the potential difficulty of choosing an appropriate acceptance rate. The main drawback of the proposed approach concerns joint parameter inference. There are many settings where the joint parameter distribution is of interest and the proposed RF approach cannot handle that. In population genetics for example, estimation of the severity and of the duration of the bottleneck should be estimated jointly because of identifiability issues. The challenge of performing joint parameter inference with RF might constitute a useful research perspective. References [1] Raynal L, Marin J-M, Pudlo P, Ribatet M, Robert CP, Estoup A. 2017. ABC random forests for Bayesian parameter inference. arXiv 1605.05537v4, https://arxiv.org/pdf/1605.05537 | ABC random forests for Bayesian parameter inference | Louis Raynal, Jean-Michel Marin, Pierre Pudlo, Mathieu Ribatet, Christian P. Robert, Arnaud Estoup | <p>This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (http:// dx.doi.org/ 10.24072/ pci.evolbiol.100036). Approximate Bayesian computation (ABC) has grown into a standard methodology that manages Bayesian in... | | Bioinformatics & Computational Biology, Evolutionary Applications, Other, Population Genetics / Genomics | Michael Blum | 2017-07-06 07:42:00 | ||
13 Nov 2017

Epidemiological trade-off between intra- and interannual scales in the evolution of aggressiveness in a local plant pathogen populationThe pace of pathogens’ adaptation to their host plantsRecommended by Benoit Moury based on reviews by Benoit Moury and 1 anonymous reviewerBecause of their shorter generation times and larger census population sizes, pathogens are usually ahead in the evolutionary race with their hosts. The risks linked to pathogen adaptation are still exacerbated in agronomy, where plant and animal populations are not freely evolving but depend on breeders and growers, and are usually highly genetically homogeneous. As a consequence, the speed of pathogen adaptation is crucial for agriculture sustainability. Unraveling the time scale required for pathogens’ adaptation to their hosts would notably greatly improve our estimation of the risks of pathogen emergence, the efficiency of disease control strategies and the design of epidemiological surveillance schemes. However, the temporal scale of pathogen evolution has received much less attention than its spatial scale [1]. In their study of a wheat fungal disease, Suffert et al. [2] reached contrasting conclusions about the pathogen adaptation depending on the time scale (intra- or inter-annual) and on the host genotype (sympatric or allopatric) considered, questioning the experimental assessment of this important problem. Suffert et al. [2] sampled two pairs of Zymoseptoria tritici (the causal agent of septoria leaf blotch) sub-populations in a bread wheat field plot, representing (i) isolates collected at the beginning or at the end of an epidemic in a single growing season (2009-2010 intra-annual sampling scale) and (ii) isolates collected from plant debris at the end of growing seasons in 2009 and in 2015 (inter-annual sampling scale). Then, they measured in controlled conditions two aggressiveness traits of the isolates of these four Z. tritici sub-populations, the latent period and the lesion size on leaves, on two wheat cultivars. One of the cultivars was considered as "sympatric" because it was at the source of the studied isolates and was predominant in the growing area before the experiment, whereas the other cultivar was considered as "allopatric" since it replaced the previous one and became predominant in the growing area during the sampling period. On the sympatric host, at the intra-annual scale, they observed a marginally-significant decrease in latent period and a significant decrease of the between-isolate variance for this trait, which are consistent with a selection of pathogen variants with an enhanced aggressiveness. In contrast, at the inter-annual scale, no difference in the mean or variance of aggressiveness trait values was observed on the sympatric host, suggesting a lack of pathogen adaptation. They interpreted the contrast between observations at the two time scales as the consequence of a trade-off for the pathogen between a gain of aggressiveness after several generations of asexual reproduction at the intra-annual scale and a decrease of the probability to reproduce sexually and to be transmitted from one growing season to the next. Indeed, at the end of the growing season, the most aggressive isolates are located on the upper leaves of plants, where the pathogen density and hence probably also the probability to reproduce sexually, is lower. On the allopatric host, the conclusion about the pathogen stability at the inter-annual scale was somewhat different, since a significant increase in the mean lesion size was observed (isolates corresponding to the intra-annual scale were not checked on the allopatric host). This shows the possibility for the pathogen to evolve at the inter-annual scale, for a given aggressiveness trait and on a given host. In conclusion, Suffert et al.’s [2] study emphasizes the importance of the experimental design in terms of sampling time scale and host genotype choice to analyze the pathogen adaptation to its host plants. It provides also an interesting scenario, at the crossroad of the pathogen’s reproduction regime, niche partitioning and epidemiological processes, to interpret these contrasted results. Pathogen adaptation to plant cultivars with major-effect resistance genes is usually fast, including in the wheat-Z. tritici system [3]. Therefore, this study will be of great help for future studies on pathogen adaptation to plant partial resistance genes and on strategies of deployment of such resistance at the landscape scale. References [2] Suffert F, Goyeau H, Sache I, Carpentier F, Gelisse S, Morais D and Delestre G. 2017. Epidemiological trade-off between intra- and interannual scales in the evolution of aggressiveness in a local plant pathogen population. bioRxiv, 151068, ver. 3 of 12th November 2017. doi: 10.1101/151068 [3] Brown JKM, Chartrain L, Lasserre-Zuber P and Saintenac C. 2015. Genetics of resistance to Zymoseptoria tritici and applications to wheat breeding. Fungal Genetics and Biology, 79: 33–41. doi: 10.1016/j.fgb.2015.04.017 | Epidemiological trade-off between intra- and interannual scales in the evolution of aggressiveness in a local plant pathogen population | Frederic Suffert, Henriette Goyeau, Ivan Sache, Florence Carpentier, Sandrine Gelisse, David Morais, Ghislain Delestre | The efficiency of plant resistance to fungal pathogen populations is expected to decrease over time, due to its evolution with an increase in the frequency of virulent or highly aggressive strains. This dynamics may differ depending on the scale i... | | Adaptation, Evolutionary Applications, Evolutionary Epidemiology | Benoit Moury | 2017-06-23 21:04:54 | ||
10 Nov 2017

POSTPRINT
Rates of Molecular Evolution Suggest Natural History of Life History Traits and a Post-K-Pg Nocturnal Bottleneck of PlacentalsA new approach to DNA-aided ancestral trait reconstruction in mammalsRecommended by Nicolas Galtier and Belinda Chang
Reconstructing ancestral character states is an exciting but difficult problem. The fossil record carries a great deal of information, but it is incomplete and not always easy to connect to data from modern species. Alternatively, ancestral states can be estimated by modelling trait evolution across a phylogeny, and fitting to values observed in extant species. This approach, however, is heavily dependent on the underlying assumptions, and typically results in wide confidence intervals. An alternative approach is to gain information on ancestral character states from DNA sequence data. This can be done directly when the trait of interest is known to be determined by a single, or a small number, of major effect genes. In some of these cases it can even be possible to investigate an ancestral trait of interest by inferring and resurrecting ancestral sequences in the laboratory. Examples where this has been successfully used to address evolutionary questions range from the nocturnality of early mammals [1], to the loss of functional uricases in primates, leading to high rates of gout, obesity and hypertension in present day humans [2]. Another possibility is to rely on correlations between species traits and the genome average substitution rate/process. For instance, it is well established that the ratio of nonsynonymous to synonymous substitution rate, dN/dS, is generally higher in large than in small species of mammals, presumably due to a reduced effective population size in the former. By estimating ancestral dN/dS, one can therefore gain information on ancestral body mass (e.g. [3-4]). The interesting paper by Wu et al. [5] further develops this second possibility of incorporating information on rate variation derived from genomic data in the estimation of ancestral traits. The authors analyse a large set of 1185 genes in 89 species of mammals, without any prior information on gene function. The substitution rate is estimated for each gene and each branch of the mammalian tree, and taken as an indicator of the selective constraint applying to a specific gene in a specific lineage – more constraint, slower evolution. Rate variation is modelled as resulting from a gene effect, a branch effect, and a gene X branch interaction effect, which captures lineage-specific peculiarities in the distribution of functional constraint across genes. The interaction term in terminal branches is regressed to observed trait values, and the relationship is used to predict ancestral traits from interaction terms in internal branches. The power and accuracy of the estimates are convincingly assessed via cross validation. Using this method, the authors were also able to use an unbiased approach to determine which genes were the main contributors to the evolution of the life-history traits they reconstructed. The ancestors to current placental mammals are predicted to have been insectivorous - meaning that the estimated distribution of selective constraint across genes in basal branches of the tree resembles that of extant insectivorous taxa - consistent with the mainstream palaeontological hypothesis. Another interesting result is the prediction that only nocturnal lineages have passed the Cretaceous/Tertiary boundary, so that the ancestors of current orders of placentals would all have been nocturnal. This suggests that the so-called "nocturnal bottleneck hypothesis" should probably be amended. Similar reconstructions are achieved for seasonality, sociality and monogamy – with variable levels of uncertainty. The beauty of the approach is to analyse the variance, not only the mean, of substitution rate across genes, and their methods allow for the identification of the genes contributing to trait evolution without relying on functional annotations. This paper only analyses discrete traits, but the framework can probably be extended to continuous traits as well. References [1] Bickelmann C, Morrow JM, Du J, Schott RK, van Hazel I, Lim S, Müller J, Chang BSW, 2015. The molecular origin and evolution of dim-light vision in mammals. Evolution 69: 2995-3003. doi: https://doi.org/10.1111/evo.12794 [2] Kratzer, JT, Lanaspa MA, Murphy MN, Cicerchi C, Graves CL, Tipton PA, Ortlund EA, Johnson RJ, Gaucher EA, 2014. Evolutionary history and metabolic insights of ancient mammalian uricases. Proceedings of the National Academy of Science, USA 111:3763-3768. doi: https://doi.org/10.1073/pnas.1320393111 [3] Lartillot N, Delsuc F. 2012. Joint reconstruction of divergence times and life-history evolution in placental mammals using a phylogenetic covariance model. Evolution 66:1773-1787. doi: https://doi.org/10.1111/j.1558-5646.2011.01558.x [4] Romiguier J, Ranwez V, Douzery EJ, Galtier N. 2013. Genomic evidence for large, long-lived ancestors to placental mammals. Molecular Biology and Evolution 30:5-13. doi: https://doi.org/10.1093/molbev/mss211 [5] Wu J, Yonezawa T, Kishino H. 2016. Rates of Molecular Evolution Suggest Natural History of Life History Traits and a Post-K-Pg Nocturnal Bottleneck of Placentals. Current Biology 27: 3025-3033. doi: https://doi.org/10.1016/j.cub.2017.08.043 | Rates of Molecular Evolution Suggest Natural History of Life History Traits and a Post-K-Pg Nocturnal Bottleneck of Placentals | Wu J, Yonezawa T, Kishino H. | Life history and behavioral traits are often difficult to discern from the fossil record, but evolutionary rates of genes and their changes over time can be inferred from extant genomic data. Under the neutral theory, molecular evolutionary rate i... | | Bioinformatics & Computational Biology, Life History, Molecular Evolution, Paleontology, Phylogenetics / Phylogenomics | Nicolas Galtier | 2017-11-10 14:52:26 | ||
07 Nov 2017

MaxTiC: Fast ranking of a phylogenetic tree by Maximum Time Consistency with lateral gene transfersDating nodes in a phylogeny using inferred horizontal gene transfersRecommended by Tatiana Giraud and Toni Gabaldon based on reviews by Alexandros Stamatakis, Mukul Bansal and 2 anonymous reviewersDating nodes in a phylogeny is an important problem in evolution and is typically performed by using molecular clocks and fossil age estimates [1]. The manuscript by Chauve et al. [2] reports a novel method, which uses lateral gene transfers to help ordering nodes in a species tree. The idea is that a lateral gene transfer can only occur between two species living at the same time, which indirectly informs on node relative ages in a phylogeny: the donor species cannot be more recent than the recipient species. Horizontal gene transfers are increasingly recognized as frequent, even in eukaryotes, and especially in micro-organisms that have little fossil records [3-7]. Yet, such an important source of information has been very rarely used so far for inferring relative node ages in phylogenies. In this context, the method by Chauve et al. [2] represents an innovative and original approach to a difficult problem. An obvious limitation of the approach is that it relies on inferences of horizontal transfers, which detection is in itself a difficult problem. Incomplete taxon sampling, or the extinction of the true donor lineage may render patterns difficult to interpret in a temporary fashion. Yet, for clades with no fossils this may be the only piece of information we have at hand, and the growing amount of sequence data is likely to minimize issues derived from incomplete sampling. The developed method, MaxTiC (for Maximal Time Consistency) [2], represents a very nice application of theoretical developments on the well-known « Feedback Arc Set » computer science problem to the evolutionary question of ordering nodes in a phylogeny. MaxTiC uses as input a species tree and a set of time constraints based on lateral gene transfers inferred using other softwares, and minimizes conflicts between node ordering and these time constraints. The application of MaxTiC on simulated datasets indicated that node ordering was fairly accurate [2]. MaxTiC is implemented in a freely available software, which represents original and relevant contribution to the field of evolutionary biology. References [1] Donoghue P and Smith M, editors. 2003. Telling the evolutionary time. CRC press. [2] Chauve C, Rafiey A, Davin AA, Scornavacca C, Veber P, Boussau B, Szöllősi GJ, Daubin V and Tannier E. 2017. MaxTiC: Fast ranking of a phylogenetic tree by Maximum Time Consistency with lateral gene transfers. bioRxiv 127548, ver. 6 of 6th November 2017. doi: 10.1101/127548 [3] Ropars J, Rodríguez de la Vega RC, Lopez-Villavicencio M, Gouzy J, Sallet E, Debuchy R, Dupont J, Branca A and Giraud T. 2015. Adaptive horizontal gene transfers between multiple cheese-associated fungi. Current Biology 19, 2562–2569. doi: 10.1016/j.cub.2015.08.025 [4] Novo M, Bigey F, Beyne E, Galeote V, Gavory F, Mallet S, Cambon B, Legras JL, Wincker P, Casaregola S and Dequin S. 2009. Eukaryote-to-eukaryote gene transfer events revealed by the genome sequence of the wine yeast Saccharomyces cerevisiae EC1118. Proceeding of the National Academy of Science USA, 106, 16333–16338. doi: 10.1073/pnas.0904673106 [5] Naranjo-Ortíz MA, Brock M, Brunke S, Hube B, Marcet-Houben M, Gabaldón T. 2016. Widespread inter- and intra-domain horizontal gene transfer of d-amino acid metabolism enzymes in Eukaryotes. Frontiers in Microbiology 7, 2001. doi: 10.3389/fmicb.2016.02001 [6] Alexander WG, Wisecaver JH, Rokas A, Hittinger CT. 2016. Horizontally acquired genes in early-diverging pathogenic fungi enable the use of host nucleosides and nucleotides. Proceeding of the National Academy of Science USA. 113, 4116–4121. doi: 10.1073/pnas.1517242113 [7] Marcet-Houben M, Gabaldón T. 2010. Acquisition of prokaryotic genes by fungal genomes. Trends in Genetics. 26, 5–8. doi: 10.1016/j.tig.2009.11.007 | MaxTiC: Fast ranking of a phylogenetic tree by Maximum Time Consistency with lateral gene transfers | Cédric Chauve, Akbar Rafiey, Adrian A. Davin, Celine Scornavacca, Philippe Veber, Bastien Boussau, Gergely J Szöllosi, Vincent Daubin, and Eric Tannier | Lateral gene transfers (LGTs) between ancient species contain information about the relative timing of species diversification. Specifically, the ancestors of a donor species must have existed before the descendants of the recipient species. Hence... | | Bioinformatics & Computational Biology, Evolutionary Dynamics, Genome Evolution, Life History, Molecular Evolution, Phylogenetics / Phylogenomics | Tatiana Giraud | 2017-06-28 13:40:52 | ||
06 Oct 2017

Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebratesCombining molecular information on chromatin organisation with eQTLs and evolutionary conservation provides strong candidates for the evolution of gene regulation in mammalian brainsRecommended by Marc Robinson-Rechavi based on reviews by Marc Robinson-Rechavi and Charles Danko
In this manuscript [1], Francisco J. Novo proposes candidate non-coding genomic elements regulating neurodevelopmental genes. What is very nice about this study is the way in which public molecular data, including physical interaction data, is used to leverage recent advances in our understanding to molecular mechanisms of gene regulation in an evolutionary context. More specifically, evolutionarily conserved non coding sequences are combined with enhancers from the FANTOM5 project, DNAse hypersensitive sites, chromatin segmentation, ChIP-seq of transcription factors and of p300, gene expression and eQTLs from GTEx, and physical interactions from several Hi-C datasets. The candidate regulatory regions thus identified are linked to candidate regulated genes, and the author shows their potential implication in brain development. While the results are focused on a small number of genes, this allows to verify features of these candidates in great detail. This study shows how functional genomics is increasingly allowing us to fulfill the promises of Evo-Devo: understanding the molecular mechanisms of conservation and differences in morphology. References [1] Novo, FJ. 2017. Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebrates. bioRxiv, 150482, ver. 4 of Sept 29th, 2017. doi: 10.1101/150482 | Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebrates | Francisco J. Novo | <p>Many non-coding regulatory elements conserved in vertebrates regulate the expression of genes involved in development and play an important role in the evolution of morphology through the rewiring of developmental gene networks. Available biolo... | | Genome Evolution | Marc Robinson-Rechavi | Marc Robinson-Rechavi, Charles Danko | 2017-06-29 08:55:41 | |
05 Oct 2017

Using Connectivity To Identify Climatic Drivers Of Local AdaptationA new approach to identifying drivers of local adaptationRecommended by Ruth Arabelle Hufbauer based on reviews by Ruth Arabelle Hufbauer and Thomas LenormandLocal adaptation, the higher fitness a population achieves in its local “home” environment relative to other environments is a crucial phase in the divergence of populations, and as such both generates and maintains diversity. Local adaptation is enhanced by selection and genetic variation in the relevant traits, and decreased by gene flow and genetic drift. Demonstrating local adaptation is laborious, and is typically done with a reciprocal transplant design [1], documenting repeated geographic clines [e.g. 2, 3] also provides strong evidence of local adaptation. Even when well documented, it is often unknown which aspects of the environment impose selection. Indeed, differences in environment between different sites that are measured during studies of local adaptation explain little of the variance in the degree of local adaptation [4]. This poses a problem to population management. Given climate change and habitat destruction, understanding the environmental drivers of local adaptation can be crucially important to conducting successful assisted migration or targeted gene flow. In this manuscript, Macdonald et al. [5] propose a means of identifying which aspects of the environment select for local adaptation without conducting a reciprocal transplant experiment. The idea is that the strength of relationships between traits and environmental variables that are due to plastic responses to the environment will not be influenced by gene flow, but the strength of trait-environment relationships that are due to local adaptation should decrease with gene flow. This then can be used to reduce the somewhat arbitrary list of environmental variables on which data are available down to a targeted list more likely to drive local adaptation in specific traits. To perform such an analysis requires three things: 1) measurements of traits of interest in a species across locations, 2) an estimate of gene flow between locations, which can be replaced with a biologically meaningful estimate of how well connected those locations are from the point of view of the study species, and 3) data on climate and other environmental variables from across a species’ range, many of which are available on line. Macdonald et al. [5] demonstrate their approach using a skink (Lampropholis coggeri). They collected morphological and physiological data on individuals from multiple populations. They estimated connectivity among those locations using information on habitat suitability and dispersal potential [6], and gleaned climatic data from available databases and the literature. They find that two physiological traits, the critical minimum and maximum temperatures, show the strongest signs of local adaptation, specifically local adaptation to annual mean precipitation, precipitation of the driest quarter, and minimum annual temperature. These are then aspects of skink phenotype and skink habitats that could be explored further, or could be used to provide background information if migration efforts, for example for genetic rescue [7] were initiated. The approach laid out has the potential to spark a novel genre of research on local adaptation. It its simplest form, knowing that local adaptation is eroded by gene flow, it is intuitive to consider that if connectivity reduces the strength of the relationship between an environmental variable and a trait, that the trait might be involved in local adaptation. The approach is less intuitive than that, however – it relies not connectivity per-se, but the interaction between connectivity and different environmental variables and how that interaction alters trait-environment relationships. The authors lay out a number of useful caveats and potential areas that could use further development. It will be interesting to see how the community of evolutionary biologists responds. References [1] Blanquart F, Kaltz O, Nuismer SL and Gandon S. 2013. A practical guide to measuring local adaptation. Ecology Letters, 16: 1195-1205. doi: 10.1111/ele.12150 [2] Huey RB, Gilchrist GW, Carlson ML, Berrigan D and Serra L. 2000. Rapid evolution of a geographic cline in size in an introduced fly. Science, 287: 308-309. doi: 10.1126/science.287.5451.308 [3] Milesi P, Lenormand T, Lagneau C, Weill M and Labbé P. 2016. Relating fitness to long-term environmental variations in natura. Molecular Ecology, 25: 5483-5499. doi: 10.1111/mec.13855 [4] Hereford, J. 2009. A quantitative survey of local adaptation and fitness trade-offs. The American Naturalist 173: 579-588. doi: 10.1086/597611 [5] Macdonald SL, Llewelyn J and Phillips BL. 2017. Using connectivity to identify climatic drivers of local adaptation. bioRxiv, ver. 4 of October 4, 2017. doi: 10.1101/145169 [6] Macdonald SL, Llewelyn J, Moritz C and Phillips BL. 2017. Peripheral isolates as sources of adaptive diversity under climate change. Frontiers in Ecology and Evolution, 5:88. doi: 10.3389/fevo.2017.00088 [7] Whiteley AR, Fitzpatrick SW, Funk WC and Tallmon DA. 2015. Genetic rescue to the rescue. Trends in Ecology & Evolution, 30: 42-49. doi: 10.1016/j.tree.2014.10.009 | Using Connectivity To Identify Climatic Drivers Of Local Adaptation | Stewart L. Macdonald, John Llewelyn, Ben Phillips | Despite being able to conclusively demonstrate local adaptation, we are still often unable to objectively determine the climatic drivers of local adaptation. Given the rapid rate of global change, understanding the climatic drivers of local adapta... | | Adaptation, Evolutionary Applications | Ruth Arabelle Hufbauer | Thomas Lenormand | 2017-06-06 13:06:54 | |
29 Sep 2017

Parallel diversifications of Cremastosperma and Mosannona (Annonaceae), tropical rainforest trees tracking Neogene upheaval of the South American continentUnravelling the history of Neotropical plant diversificationRecommended by Hervé Sauquet based on reviews by Thomas Couvreur and Hervé SauquetSouth American rainforests, particularly the Tropical Andes, have been recognized as the hottest spot of plant biodiversity on Earth, while facing unprecedented threats from human impact [1,2]. Considerable research efforts have recently focused on unravelling the complex geological, bioclimatic, and biogeographic history of the region [3,4]. While many studies have addressed the question of Neotropical plant diversification using parametric methods to reconstruct ancestral areas and patterns of dispersal, Pirie et al. [5] take a distinct, complementary approach. Based on a new, near-complete molecular phylogeny of two Neotropical genera of the flowering plant family Annonaceae, the authors modelled the ecological niche of each species and reconstructed the history of niche differentiation across the region. The main conclusion is that, despite similar current distributions and close phylogenetic distance, the two genera experienced rather distinct processes of diversification, responding differently to the major geological events marking the history of the region in the last 20 million years (Andean uplift, drainage of Lake Pebas, and closure of the Panama Isthmus). As a researcher who has not personally worked on Neotropical biogeography, I found this paper captivating and especially enjoyed very much reading the Introduction, which sets out the questions very clearly. The strength of this paper is the near-complete diversity of species the authors were able to sample in each clade and the high-quality data compiled for the niche models. I would recommend this paper as a nice example of a phylogenetic study aimed at unravelling the detailed history of Neotropical plant diversification. While large, synthetic meta-analyses of many clades should continue to seek general patterns [4,6], careful studies restricted on smaller, but well controlled and sampled datasets such as this one are essential to really understand tropical plant diversification in all its complexity. References [1] Antonelli A, and Sanmartín I. 2011. Why are there so many plant species in the Neotropics? Taxon 60, 403–414. [2] Mittermeier RA, Robles-Gil P, Hoffmann M, Pilgrim JD, Brooks TB, Mittermeier CG, Lamoreux JL and Fonseca GAB. 2004. Hotspots revisited: Earths biologically richest and most endangered ecoregions. CEMEX, Mexico City, Mexico 390pp [3] Antonelli A, Nylander JAA, Persson C and Sanmartín I. 2009. Tracing the impact of the Andean uplift on Neotropical plant evolution. Proceedings of the National Academy of Science of the USA 106, 9749–9754. doi: 10.1073/pnas.0811421106 [4] Hoorn C, Wesselingh FP, ter Steege H, Bermudez MA, Mora A, Sevink J, Sanmartín I, Sanchez-Meseguer A, Anderson CL, Figueiredo JP, Jaramillo C, Riff D, Negri FR, Hooghiemstra H, Lundberg J, Stadler T, Särkinen T and Antonelli A. 2010. Amazonia through time: Andean uplift, climate change, landscape evolution, and biodiversity. Science 330, 927–931. doi: 10.1126/science.1194585 [5] Pirie MD, Maas PJM, Wilschut R, Melchers-Sharrott H and Chatrou L. 2017. Parallel diversifications of Cremastosperma and Mosannona (Annonaceae), tropical rainforest trees tracking Neogene upheaval of the South American continent. bioRxiv, 141127, ver. 3 of 28th Sept 2017. doi: 10.1101/141127 [6] Bacon CD, Silvestro D, Jaramillo C, Tilston Smith B, Chakrabartye P and Antonelli A. 2015. Biological evidence supports an early and complex emergence of the Isthmus of Panama. Proceedings of the National Academy of Science of the USA 112, 6110–6115. doi: 10.1073/pnas.1423853112 | Parallel diversifications of Cremastosperma and Mosannona (Annonaceae), tropical rainforest trees tracking Neogene upheaval of the South American continent | Michael D. Pirie, Paul J. M. Maas, Rutger A. Wilschut, Heleen Melchers-Sharrott & Lars W. Chatrou | Much of the immense present day biological diversity of Neotropical rainforests originated from the Miocene onwards, a period of geological and ecological upheaval in South America. We assess the impact of the Andean orogeny, drainage of lake Peba... | | Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Hervé Sauquet | Hervé Sauquet, Thomas Couvreur | 2017-06-03 21:25:48 |




