Tell me who you mate with, I’ll tell you what’s going on
Recommended by Sara Magalhaes and Alexandre Courtiol based on reviews by Alexandre Courtiol and 2 anonymous reviewers
The study of sexual selection goes as far as Darwin himself. Since then, elaborate theories concerning both intra- and inter-sexual sexual have been developed, and elegant experiments have been designed to test this body of theory. It may thus come as a surprise that the community is still debating on the correct way to measure simple components of sexual selection, such as the Bateman gradient (i.e., the covariance between the number of matings and the number of offspring)[1,2], or to quantify complex behaviours such as mate choice (the non-random choice of individuals with particular characters as mates)[3,4] and their consequences.
One difficulty in the study of sexual selection is evaluating the consequences of non-random mating. Indeed, when non-random mating is observed in a population, it is often difficult to establish whether such mating pattern leads to i) sexual selection per se (selection pressures favouring certain phenotypes), and/or ii) the non-random association of parental genes in their offspring or not. These two processes differ. In particular, assortative (and disassortative) mating can shape genetic covariances without leading to changes in gene frequencies in the population. Their distinction matters because these two processes lead to different evolutionary outcomes, which can have large ripple effects in the evolution of sexual behaviours, sexual ornamentation, and speciation.
In his paper, entitled “Multi-model inference of non-random mating from an information theoretic approach” [5], Carvajal-Rodríguez tackled this issue. The author generated a simple model in which the consequences of non-random mating can be inferred from information on the population frequencies before and after mating. The procedure is as follows: from the initial population frequencies of phenotypes (or genotypes) of both sexes, the model generates predictions on the frequencies after mating, assuming that particular mating patterns have occurred. This leads to different predictions for the phenotypic (or genotypic) frequencies after mating. The particular mating pattern leading to the best fit with the real frequencies is then identified via a model selection procedure (performing model averaging to combine different mating patterns is also possible).
This study builds on a framework introduced by Carvajal-Rodríguez’s colleagues [6] and encompasses later methodological developments involving the author himself [7]. Compared to early work, the new method proposed by the author builds on the relationship between mating pattern and information [8] to distinguish among scenarios that would lead to non-random mating due to different underlying processes, using simple model selection criterion such as the AICc.
The great asset of the proposed method is that it can be applied to the study of natural populations in which the study of mate choice and sexual selection is notoriously difficult. In the manuscript, the procedure is tested on a population of marine gastropods (Littorina saxatilis). This allows the reader to grasp how the method can be applied to a real system. In fact, anyone can try out the method thanks to the freely available software InfoMating programmed by the author. One important assumption underlying the current method is that the frequencies of unmated individuals do not change during the mating season. If this is not the case, the reader may refer to another publication of the same author which relaxes this assumption [9]. These papers are both instrumental for empiricists interested in testing sexual selection theory.
References
[1] Bateman, A. J. (1948). Intra-sexual selection in Drosophila. Heredity, 2(3), 349-368. doi: 10.1038/hdy.1948.21
[2] Jones, A. G. (2009). On the opportunity for sexual selection, the Bateman gradient and the maximum intensity of sexual selection. Evolution: International Journal of Organic Evolution, 63(7), 1673-1684. doi: 10.1111/j.1558-5646.2009.00664.x
[3] Andersson, M., & Simmons, L. W. (2006). Sexual selection and mate choice. Trends in ecology & evolution, 21(6), 296-302. doi: 10.1016/j.tree.2006.03.015
[4] Kuijper, B., Pen, I., & Weissing, F. J. (2012). A guide to sexual selection theory. Annual Review of Ecology, Evolution, and Systematics, 43, 287-311. doi: 10.1146/annurev-ecolsys-110411-160245
[5] Carvajal-Rodríguez, A. (2019). Multi-model inference of non-random mating from an information theoretic approach. bioRxiv, 305730, ver. 5 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/305730
[6] Rolán‐Alvarez, E., & Caballero, A. (2000). Estimating sexual selection and sexual isolation effects from mating frequencies. Evolution, 54(1), 30-36. doi: 10.1111/j.0014-3820.2000.tb00004.x
[7] Carvajal-Rodríguez, A., & Rolan-Alvarez, E. (2006). JMATING: a software for the analysis of sexual selection and sexual isolation effects from mating frequency data. BMC Evolutionary Biology, 6(1), 40. doi: 10.1186/1471-2148-6-40
[8] Carvajal-Rodríguez, A. (2018). Non-random mating and information theory. Theoretical population biology, 120, 103-113. doi: 10.1016/j.tpb.2018.01.003
[9] Carvajal-Rodríguez, A. (2019). A generalization of the informational view of non-random mating: Models with variable population frequencies. Theoretical population biology, 125, 67-74. doi: 10.1016/j.tpb.2018.12.004
| Multi-model inference of non-random mating from an information theoretic approach | Antonio Carvajal-Rodríguez | <p>Non-random mating has a significant impact on the evolution of organisms. Here, I developed a modelling framework for discrete traits (with any number of phenotypes) to explore different models connecting the non-random mating causes (mate comp... | 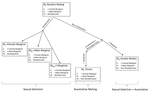 | Evolutionary Ecology, Evolutionary Theory, Sexual Selection | Sara Magalhaes | | 2019-02-08 19:24:03 | View |
Thermal regimes, but not mean temperatures, drive patterns of rapid climate adaptation at a continent-scale: evidence from the introduced European earwig across North America
Jean-Claude Tourneur, Joël Meunier
https://doi.org/10.1101/550319
Temperature variance, rather than mean, drives adaptation to local climate
Recommended by Fabien Aubret based on reviews by Ben Phillips and Eric Gangloff
Climate change is impacting eco-systems worldwide and driving many populations to move, adapt or go extinct. It is increasingly appreciated, for example, that species may adjust their phenology in response to climate change, although empirical data is scarce. In this preprint [1], Tourneur and Meunier report an impressive sampling effort in which life-history traits were measured across introduced populations of earwig in North America. The authors examine whether variation in life-history across populations is correlated with aspects of the thermal climate experienced by each population: mean temperature and seasonality of temperature. They find some fascinating correlations between life-history and thermal climate; correlations with the seasonality of temperature, but not with mean temperature. This study provides relatively uncommon data, in the sense that where most of the literature looking at adaptation in animals in response to climate change has focused on physiological traits [2, 3], this study examines changes in life-history traits with time scales relevant to impending climate change, and provides a reasonable argument that this is adaptation, not just constraint.
References
[1] Tourneur, J.-C. and Meunier, J. (2019). Thermal regimes, but not mean temperatures, drive patterns of rapid climate adaptation at a continent-scale: evidence from the introduced European earwig across North America. BioRxiv, 550319, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/550319
[2] Kellermann, V., Overgaard, J., Hoffmann, A. A., Fløjgaard, C., Svenning, J. C., & Loeschcke, V. (2012). Upper thermal limits of Drosophila are linked to species distributions and strongly constrained phylogenetically. Proceedings of the National Academy of Sciences, 109(40), 16228-16233. doi: 10.1073/pnas.1207553109
[3] Hoffmann, A. A., & Sgro, C. M. (2011). Climate change and evolutionary adaptation. Nature, 470(7335), 479. doi: 10.1038/nature09670
| Thermal regimes, but not mean temperatures, drive patterns of rapid climate adaptation at a continent-scale: evidence from the introduced European earwig across North America | Jean-Claude Tourneur, Joël Meunier | <p>The recent development of human societies has led to major, rapid and often inexorable changes in the environment of most animal species. Over the last decades, a growing number of studies formulated predictions on the modalities of animal adap... |  | Adaptation, Evolutionary Ecology, Life History | Fabien Aubret | | 2019-02-15 09:12:11 | View |
Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network
Maud Irène Tenaillon, Khawla Sedikki, Maeva Mollion, Martine Le Guilloux, Elodie Marchadier, Adrienne Ressayre, Christine Dillmann
https://doi.org/10.1101/461947
Early and late flowering gene expression patterns in maize
Recommended by Tanja Pyhäjärvi based on reviews by Laura Shannon ? and 2 anonymous reviewers
Artificial selection experiments are key experiments in evolutionary biology. The demonstration that application of selective pressure across multiple generations results in heritable phenotypic changes is a tangible and reproducible proof of the evolution by natural selection.
Artificial selection experiments are used to evaluate the joint effects of selection on multiple traits, their genetic covariances and differences in responses in different environments. Most studies on artificial selection experiments report and base their analyses on phenotypic changes [1]. More recently, changes in allele frequency and other patterns of molecular genetic diversity have been used to identify genomic locations where selection has had an effect. However, so far the changes in gene expression have not been in the focus of artificial selection experiment studies (see [2] for an example though).
In plants, one of the most famous artificial selection experiments is the Illinois Corn Experiment where maize (Zea mays) is selected for oil and protein content [3], but in addition, similar experiments have been conducted also for other traits in maize. In Saclay divergent selection experiment [4] two maize inbred lines (F252 and MBS847) have been selected for early and late flowering for 13 generations, resulting in two week difference in flowering time.
In ”Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network ” [5] Maud Tenaillon and her coworkers study the gene expression differences among these two independently selected maize populations. Their experiments cover two years in field conditions and they use samples of shoot apical meristem at three different developmental stages: vegetative, transitioning and reproductive. They use RNA-seq transcriptome level differences and qRT-PCR for gene expression pattern investigation. The work is continuation to earlier genetic and phenotypic studies on the same material [4, 6].
The reviewers and I agree that dataset is unique and its major benefit is that it has been obtained from field conditions similar to those that species may face under natural setting during selection. Their tissue sampling is supported by flowering time phenotypic observations and covers the developmental transition stage, making a good effort to identify key transcriptional and phenotypic changes and their timing affected by selection.
Tenaillon et al. [5] identify more than 2000 genes that are differentially expressed among early and late flowering populations. Expectedly, they are enriched for known flowering time genes. As they point out, differential expression of thousands of genes does not mean that they all were independently affected by selection, but rather that the whole transcriptional network has shifted, possibly due to just few upstream or hub-genes. Also, the year-to-year variation had smaller effect in gene expression compared to developmental stage or genetic background, possibly indicating selection for stability across environmental fluctuation for such an important phenotype as flowering time.
Another noteworthy observation is that they find convergent patterns of transcriptional changes among the two selected lines. 115 genes expression patterns are shifted due to selection in both genetic backgrounds. This convergent pattern can be a result of either selection on standing variation or de novo mutations. The data does not allow testing which process is underlying the observed convergence. However, their results show that this is an interesting future question that can be addressed using genotype and gene expression data from the same ancestral and derived material and possibly their hybrids.
References
[1] Hill, W. G., & Caballero, A. (1992). Artificial selection experiments. Annual Review of Ecology and Systematics, 23(1), 287-310. doi: 10.1146/annurev.es.23.110192.001443
[2] Konczal, M., Babik, W., Radwan, J., Sadowska, E. T., & Koteja, P. (2015). Initial molecular-level response to artificial selection for increased aerobic metabolism occurs primarily through changes in gene expression. Molecular biology and evolution, 32(6), 1461-1473. doi: 10.1093/molbev/msv038
[3] Moose, S. P., Dudley, J. W., & Rocheford, T. R. (2004). Maize selection passes the century mark: a unique resource for 21st century genomics. Trends in plant science, 9(7), 358-364. doi: 10.1016/j.tplants.2004.05.005
[4] Durand, E., Tenaillon, M. I., Ridel, C., Coubriche, D., Jamin, P., Jouanne, S., Ressayre, A., Charcosset, A. and Dillmann, C. (2010). Standing variation and new mutations both contribute to a fast response to selection for flowering time in maize inbreds. BMC evolutionary biology, 10(1), 2. doi: 10.1186/1471-2148-10-2
[5] Tenaillon, M. I., Seddiki, K., Mollion, M., Le Guilloux, M., Marchadier, E., Ressayre, A. and Dillmann C. (2019). Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network. BioRxiv, 461947 ver. 5 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/461947
[6] Durand, E., Tenaillon, M. I., Raffoux, X., Thépot, S., Falque, M., Jamin, P., Bourgais A., Ressayre, A. and Dillmann, C. (2015). Dearth of polymorphism associated with a sustained response to selection for flowering time in maize. BMC evolutionary biology, 15(1), 103. doi: 10.1186/s12862-015-0382-5
| Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network | Maud Irène Tenaillon, Khawla Sedikki, Maeva Mollion, Martine Le Guilloux, Elodie Marchadier, Adrienne Ressayre, Christine Dillmann | <p>Artificial selection experiments are designed to investigate phenotypic evolution of complex traits and its genetic basis. Here we focused on flowering time, a trait of key importance for plant adaptation and life-cycle shifts. We undertook div... |  | Adaptation, Experimental Evolution, Expression Studies, Quantitative Genetics | Tanja Pyhäjärvi | | 2018-11-23 11:57:35 | View |
When sinks become sources: adaptive colonization in asexuals
Florian Lavigne, Guillaume Martin, Yoann Anciaux, Julien Papaïx, Lionel Roques
https://doi.org/10.1101/433235
Fisher to the rescue
Recommended by François Blanquart and Florence Débarre based on reviews by 3 anonymous reviewers
The ability of a population to adapt to a new niche is an important phenomenon in evolutionary biology. The colonisation of a new volcanic island by plant species; the colonisation of a host treated by antibiotics by a-resistant strain; the Ebola virus transmitting from bats to humans and spreading epidemically in Western Africa, are all examples of a population invading a new niche, adapting and eventually establishing in this new environment.
Adaptation to a new niche can be studied using source-sink models. In the original environment —the “source”—, the population enjoys a positive growth-rate and is self-sustaining, while in the new environment —the “sink”— the population has a negative growth rate and is able to sustain only by the continuous influx of migrants from the source. Understanding the dynamics of adaptation to the sink environment is challenging from a theoretical standpoint, because it requires modelling the demography of the sink as well as the transient dynamics of adaptation. Moreover, local selection in the sink and immigration from the source create distributions of genotypes that complicate the use of many common mathematical approaches.
In their paper, Lavigne et al. [1], develop a new deterministic model of adaptation to a harsh sink environment in an asexual species. The fitness of an individual is maximal when a number of phenotypes are tuned to an optimal value, and declines monotonously as phenotypes are further away from this optimum. This model —called Fisher’s Geometric Model— generates a GxE interaction for fitness because the phenotypic optimum in the sink environment is distinct from that in the source environment [2]. The authors circumvent mathematical difficulties by developing an original approach based on tracking the deterministic dynamics of the cumulant generating function of the fitness distribution in the sink. They derive a number of important results on the dynamics of adaptation to the sink:
From the point where immigration from the source to the sink starts, four phases of adaptation are observed. After a short transient phase (phase 1), a migration-selection balance is reached in the sink (phase 2). After a while, thanks to the immigration of rare adapted migrants and mutation in the sink, a small fraction of the sink population exhibits a close-to-optimal phenotype. This small adapted fraction grows in frequency and mean fitness rapidly increases in the sink (phase 3). Finally, the population settles around the sink optimum (phase 4) and, hurray, the sink is now a source! Interestingly, in this model the evolutionary dynamics do not depend on the immigration rate. In other words, adaptation will proceed at the same rate regardless of how many immigrants invade the sink. This is because the impact of immigration on adaptation depends on the rate of immigration relative to the sink density. This ratio is actually independent of immigration in a model where the sink is initially empty, migration from the sink back to the source is negligible and without density-dependence in the sink. In this model, mutation is a double-edged sword. Adapted phenotypes emerge from new mutations, and under this effect alone a higher mutation rate would translate into a shorter time to establishment in the sink. However, mutations may also have deleterious effects by displacing the phenotype away from the optimum. This mutation load will be greater when individuals need to simultaneously tune a large number of phenotypes. As a consequence of these two effects of mutations, time to establishment is minimal for an intermediate mutation rate. This result emerges from Fisher’s Geometric Model, but may hold more generally for biologically plausible fitness landscapes where mutations generates both beneficial (allowing adaptation to the sink) and deleterious genotypes. Lastly, in Fisher’s Geometric Model, the time to establishment increases superlinearly with harshness of the sink when the sink is too harsh, and establishment may occur only after a very long time. In these harsh sinks, the adapted genotypes are very few and increase very slowly in frequency, making the second phase of adaptation much longer. Thus, and as a direct consequence of Fisher’s Geometric Model, adding a “stepping stone” intermediate environment would allow faster adaptation to the extreme environment.
In conclusion, this theoretical work presents a method based on Fisher’s Geometric Model and the use of cumulant generating functions to resolve some aspects of adaptation to a sink environment. It generates a number of theoretical predictions for the adaptive colonisation of a sink by an asexual species with some standing genetic variation. It will be a fascinating task to examine whether these predictions hold in experimental evolution systems: will we observe the four phases of the dynamics of mean fitness in the sink environment? Will the rate of adaptation indeed be independent of the immigration rate? Is there an optimal rate of mutation for adaptation to the sink? Such critical tests of the theory will greatly improve our understanding of adaptation to novel environments.
References
[1] Lavigne, F., Martin, G., Anciaux, Y., Papaïx, J., and Roques, L. (2019). When sinks become sources: adaptive colonization in asexuals. bioRxiv, 433235, ver. 5 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/433235
[2] Martin, G., and Lenormand, T. (2006). A general multivariate extension of Fisher's geometrical model and the distribution of mutation fitness effects across species. Evolution, 60, 893-907. doi: 10.1111/j.0014-3820.2006.tb01169.x
| When sinks become sources: adaptive colonization in asexuals | Florian Lavigne, Guillaume Martin, Yoann Anciaux, Julien Papaïx, Lionel Roques | <p>The successful establishment of a population into a new empty habitat outside of its initial niche is a phenomenon akin to evolutionary rescue in the presence of immigration. It underlies a wide range of processes, such as biological invasions ... |  | Adaptation, Evolutionary Applications, Evolutionary Dynamics, Evolutionary Ecology | François Blanquart | | 2018-10-03 20:59:16 | View |
One (more) step towards a dynamic view of the Latitudinal Diversity Gradient
Recommended by Joaquín Hortal and Juan Arroyo based on reviews by Juan Arroyo, Joaquín Hortal, Arne Mooers, Joaquin Calatayud and 2 anonymous reviewers based on reviews by Juan Arroyo, Joaquín Hortal, Arne Mooers, Joaquin Calatayud and 2 anonymous reviewers
The Latitudinal Diversity Gradient (LDG) has fascinated natural historians, ecologists and evolutionary biologists ever since [1] described it about 200 years ago [2]. Despite such interest, agreement on the origin and nature of this gradient has been elusive. Several tens of hypotheses and models have been put forward as explanations for the LDG [2-3], that can be grouped in ecological, evolutionary and historical explanations [4] (see also [5]). These explanations can be reduced to no less than 26 hypotheses, which account for variations in ecological limits for the establishment of progressively larger assemblages, diversification rates, and time for species accumulation [5]. Besides that, although in general the tropics hold more species, different taxa show different shapes and rates of spatial variation [6], and a considerable number of groups show reverse patterns, with richer assemblages in cold temperate regions (see e.g. [7-9]).
Understanding such complexity needs integrating ecological and evolutionary research into the wide temporal and spatial perspectives provided by the burgeoning field of biogeography. This integrative discipline ¬–that traces back to Humboldt himself (e.g. [10])– seeks to put together historical and functional explanations to explain the complex dynamics of Earth’s biodiversity. Different to quantum physicists, biogeographers cannot pursue the ultimate principle behind the patterns we observe in nature due to the interplay of causes and effects, which in fact tell us that there is not such a single principle. Rather, they need to identify an array of basic principles coming from different perspectives, to then integrate them into models that provide realistic –but never simple– explanations to biodiversity gradients such as LDG (see, e.g., [5; 11]). That is, rather than searching for a sole explanation, research on the LDG must aim to identify as many signals hidden in the pattern as possible, and provide hypotheses or models that account for these signals. To later integrate them and, whenever possible, to validate them with empirical data on the organisms’ distribution, ecology and traits, phylogenies, fossils, etc.
Within this context, Meseguer & Condamine [12] provide a novel perspective to LDG research using phylogenetic and fossil evidence on the origin and extinction of taxa within the turtle, crocodile and lizard (i.e. squamate) lineages. By digging into deep time down to the Triassic (about 250 Myr ago) they are able to identify several episodes of flattening and steepening of the LDG for these three clades. Strikingly, their results show similar diversification rates in the northern hemisphere and in the equator during the over 100 Myr long global greenhouse period that extends from the late Jurassic to the Cretaceous and early Neogene. During this period, the LDG for these three groups would have appeared quite even across a mainly tropical Globe, although the equatorial regions were apparently much more evolutionarily dynamic. The equator shows much higher rates of origination and extinction of branches throughout the Cretaceous, but they counteract each other so net diversification is similar to that of the northern hemisphere in all three groups. The transition to a progressively colder Earth in the Paleogene (starting around 50 Myr ago) provokes a mass extinction in the three clades, which is compensated in the equator by the dispersal of many taxa from the areas that currently pertain to the Holarctic biogeographical realm. Finally, during the coldhouse Earth’s climatic conditions of the Neogene only squamates show significant positive diversification rates in extratropical areas, while the diversity of testudines remains, and crocodiles continue declining progressively towards oblivion in the whole world.
Meseguer & Condamine [12] attribute these temporal patterns to the so-called asymmetric gradient of extinction and dispersal (AGED) framework. Here, the dynamics of extinction-at and dispersal-from high latitudes during colder periods increase the steepness of the LDG. Whereas the gradient flattens when Earth warms up as a result of dispersal from the equator followed by increased diversification in extratropical regions. This idea in itself is not new, for the influence of climatic oscillations on diversification rates is well known, at least for the Pleistocene Ice Ages [13], as is the effect of niche conservatism on the LDG [14]. Nevertheless, Meseguer & Condamine’s AGED provides a synthetic verbal model that could allow integrating the three main types of processes behind the LDG into a single framework. To do this it would be necessary to combine AGED’s cycles of dispersal and diversification with realistic models of: (1) the ecological limits to host rich assemblages in the colder and less productive temperate climatic domains; (2) the variations in diversification rates with shifts in temperature and/or energy regimes; and (3) the geographical patterns of climatic oscillation through time that determine the time for species accumulation in each region.
Integrating these models may allow transposing Meseguer & Condamine’s [12] framework into the more mechanistic macroecological models advocated by Pontarp et al. [5]. This type of mechanistic models has been already used to understand the development of biodiversity gradients through the climatic oscillations of the Pleistocene and the Quaternary (e.g. [11]). So the challenge in this case would be to generate a realistic scenario of geographical dynamics that accounts for plate tectonics and long-term climatic oscillations. This is still a major gap and we would benefit from the integrated work by historical geologists and climatologists here. For instance, there is little doubt about the progressive cooling through the Cenozoic based in isotope recording in sea floor sediments [15]. Meseguer & Condamine [12] use this evidence for separating greenhouse, transition and coldhouse world scenarios, which should not be a problem for these rough classes. However, a detailed study of the evolutionary correlation of true climate variables across the tree of life is still pending, as temperature is inferred only for sea water in an ice-free ocean, say earlier half of the Cenozoic [15]. Precipitation regime is even less known. Such scenario would provide a scaffold upon which the temporal dynamics of several aspects of the generation and loss of biodiversity can be modelled. Additionally, one of the great advantages of selecting key clades to study the LDG would be to determine the functional basis of diversification. There are species traits that are well known to affect speciation and extinction probabilities, such as reproductive strategies or life histories (e.g. [16]). Whereas these traits might also be a somewhat redundant effect of climatic causes, they might foster (i.e. “extended reinforcement”, [17]) or slow diversification. Even so, it is unlikely that such a model would account for all the latitudinal variation in species richness. But it will at least provide a baseline for the main latitudinal variations in the diversity of the regional communities (sensu [18]) worldwide. Within this context the effects of recent ecological, evolutionary and historical processes, such as environmental heterogeneity, current diversification rates or glacial cycles, will only modify the general LDG pattern resulting from the main processes contained in Meseguer & Condamine’s AGED, thereby providing a more comprehensive understanding of the geographical gradients of diversity.
References
[1] Humboldt, A. v. (1808). Ansichten der Natur, mit wissenschaftlichen Erläuterungen. J. G. Cotta, Tübingen.
[2] Hawkins, B. A. (2001). Ecology's oldest pattern? Trends in Ecology & Evolution, 16, 470. doi: 10.1016/S0169-5347(01)02197-8
[3] Lomolino, M. V., Riddle, B. R. & Whittaker, R. J. (2017). Biogeography. Fifth Edition. Sinauer Associates, Inc., Sunderland, Massachussets.
[4] Mittelbach, G. G., Schemske, D. W., Cornell, H. V., Allen, A. P., Brown, J. M., Bush, M. B., Harrison, S. P., Hurlbert, A. H., Knowlton, N., Lessios, H. A., McCain, C. M., McCune, A. R., McDade, L. A., McPeek, M. A., Near, T. J., Price, T. D., Ricklefs, R. E., Roy, K., Sax, D. F., Schluter, D., Sobel, J. M. & Turelli, M. (2007). Evolution and the latitudinal diversity gradient: speciation, extinction and biogeography. Ecology Letters, 10, 315-331. doi: 10.1111/j.1461-0248.2007.01020.x
[5] Pontarp, M., Bunnefeld L., Cabral, J. S., Etienne, R. S., Fritz, S. A., Gillespie, R. Graham, C. H., Hagen, O., Hartig, F., Huang, S., Jansson, R., Maliet, O., Münkemüller, T., Pellissier, L., Rangel, T. F., Storch, D., Wiegand, T. & Hurlbert, A. H. (2019). The latitudinal diversity gradient: novel understanding through mechanistic eco-evolutionary models. Trends in ecology & evolution, 34, 211-223. doi: 10.1016/j.tree.2018.11.009
[6] Hillebrand, H. (2004). On the generality of the latitudinal diversity gradient. The American Naturalist, 163, 192-211. doi: 10.1086/381004
[7] Santos, A. M. C. & Quicke, D. L. J. (2011). Large-scale diversity patterns of parasitoid insects. Entomological Science, 14, 371-382. doi: 10.1111/j.1479-8298.2011.00481.x
[8] Morinière, J., Van Dam, M. H., Hawlitschek, O., Bergsten, J., Michat, M. C., Hendrich, L., Ribera, I., Toussaint, E. F. A. & Balke, M. (2016). Phylogenetic niche conservatism explains an inverse latitudinal diversity gradient in freshwater arthropods. Scientific Reports, 6, 26340. doi: 10.1038/srep26340
[9] Weiser, M. D., Swenson, N. G., Enquist, B. J., Michaletz, S. T., Waide, R. B., Zhou, J. & Kaspari, M. (2018). Taxonomic decomposition of the latitudinal gradient in species diversity of North American floras. Journal of Biogeography, 45, 418-428. doi: 10.1111/jbi.13131
[10] Humboldt, A. v. (1805). Essai sur la geographie des plantes; accompagné d'un tableau physique des régions equinoxiales. Levrault, Paris.
[11] Rangel, T. F., Edwards, N. R., Holden, P. B., Diniz-Filho, J. A. F., Gosling, W. D., Coelho, M. T. P., Cassemiro, F. A. S., Rahbek, C. & Colwell, R. K. (2018). Modeling the ecology and evolution of biodiversity: Biogeographical cradles, museums, and graves. Science, 361, eaar5452. doi: 10.1126/science.aar5452
[12] Meseguer, A. S. & Condamine, F. L. (2019). Ancient tropical extinctions contributed to the latitudinal diversity gradient. bioRxiv, 236646, ver. 4 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/236646
[13] Jansson, R., & Dynesius, M. (2002). The fate of clades in a world of recurrent climatic change: Milankovitch oscillations and evolution. Annual review of ecology and systematics, 33(1), 741-777. doi: 10.1146/annurev.ecolsys.33.010802.150520
[14] Wiens, J. J., & Donoghue, M. J. (2004). Historical biogeography, ecology and species richness. Trends in ecology & evolution, 19, 639-644. doi: 10.1016/j.tree.2004.09.011
[15] Zachos, J. C., Dickens, G. R., & Zeebe, R. E. (2008). An early Cenozoic perspective on greenhouse warming and carbon-cycle dynamics. Nature, 451, 279-283. doi: 10.1038/nature06588
[16] Zúñiga-Vega, J. J., Fuentes-G, J. A., Ossip-Drahos, A. G., & Martins, E. P. (2016). Repeated evolution of viviparity in phrynosomatid lizards constrained interspecific diversification in some life-history traits. Biology letters, 12, 20160653. doi: 10.1098/rsbl.2016.0653
[17] Butlin, R. K., & Smadja, C. M. (2018). Coupling, reinforcement, and speciation. The American Naturalist, 191, 155-172. doi: 10.1086/695136
[18] Ricklefs, R. E. (2015). Intrinsic dynamics of the regional community. Ecology letters, 18, 497-503. doi: 10.1111/ele.12431
| Ancient tropical extinctions contributed to the latitudinal diversity gradient | Andrea S. Meseguer, Fabien Condamine | <p>Biodiversity currently peaks at the equator, decreasing toward the poles. Growing fossil evidence suggest that this hump-shaped latitudinal diversity gradient (LDG) has not been persistent through time, with similar species diversity across lat... | 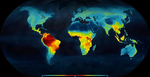 | Evolutionary Dynamics, Evolutionary Ecology, Macroevolution, Paleontology, Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Joaquín Hortal | | 2017-12-20 14:58:01 | View |
Evolution of selfing & lifespan 2.0
Recommended by Thomas Bataillon based on reviews by 2 anonymous reviewers
Flowering plants display a staggering diversity of both mating systems and life histories, ranging from almost exclusively selfers to obligate outcrossers, very short-lived annual herbs to super long lived trees. One pervasive pattern that has attracted considerable attention is the tight correlation that is found between mating systems and lifespan [1]. Until recently, theoretical explanations for these patterns have relied on static models exploring the consequences of several non-mutually exclusive important process: levels of inbreeding depression and ability to successfully were center stage. This make sense: successful colonization after long‐distance dispersal is far more likely to happen for self‐compatible than for self‐incompatible individuals in a sexually reproducing species. Furthermore, inbreeding depression (essentially a genetically driven phenomenon) and reproductive insurance are expected to shape the evolution of both mating system and lifespan.
But modelling jointly several processes and how their interplay to shape the evolution of a trait is challenging enough so models for the evolution of mating system tend invariably – for mathematical convenience and tractability – to fix lifespan [2].
However, comparative analysis of between species variations that map traits transitions among sister species in phylogenetic trees reveals a pervasive pattern: frequent transitions from a state outcrossing perennial to selfing annuals. This beg the question: is one transition triggering the other and if so, what comes first or are these transitions happening together?
In this work, Lesaffre and Billiard use a very sophisticated machinery developed by Kirkpatrick et al. [3] and consider a general class of so-called modifiers models [4]. They study jointly the evolution of life span and mating system. They do so by using models where different life stages are tracked with life stage having some (fixed for once) amount of inbreeding depression. Their paper is technically demanding, mixing analytics and computer simulations, and along the way generates several important findings that are expected to stimulate further empirical and theoretical studies: (1) pure selfing versus pure outcrossing is the expected stable evolutionary outcomes (despite observation that mixed mating systems can be regularly met in nature), (2) increasing life-span drastically reduces the scope for the evolution of selfing, conversely (3) transition to selfing will also select for shorter life span as a way to mitigate the cumulative effects of inbreeding depression on adult life stages.
As usual there is room for future work, in particular the authors’ model assumes fixed inbreeding depression in the different life stages and this highlights the need for models that explore how inbreeding depression, a pivotal quantity in these models, can itself be molded by both mating system and lifespan. A third-generation of models should be “soon” on the way!
References
[1] Grossenbacher D, Briscoe Runquist R, Goldberg EE, and Brandvain Y. (2015) Geographic range size is predicted by plant mating system. Ecology Letters 18, 706–713. doi: 10.1111/ele.12449
[2] Morgan MT, Schoen DJ, and Bataillon T. (1997) The evolution of self-fertilization in perennials. The American Naturalist 150, 618–638. doi: 10.1086/286085
[3] Kirkpatrick M, Johnson T, and Barton N. (2002) General models of multilocus evolution. Genetics 161, 1727–1750.
[4] Lesaffre, T, and Billiard S. (2019) The joint evolution of lifespan and self-fertilisation. bioRxiv, 420877, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/420877
| The joint evolution of lifespan and self-fertilisation | Thomas Lesaffre, Sylvain Billiard | <p>In Angiosperms, there exists a strong association between mating system and lifespan. Most self-fertilising species are short-lived and most predominant or obligate outcrossers are long-lived. This association is generally explained by the infl... |  | Evolutionary Theory, Life History, Reproduction and Sex | Thomas Bataillon | | 2018-09-19 10:03:51 | View |
Architectural traits constrain the evolution of unisexual flowers and sexual segregation within inflorescences: an interspecific approach
Rubén Torices, Ana Afonso, Arne A. Anderberg, José M. Gómez and Marcos Méndez
https://doi.org/10.1101/356147
Sometimes, sex is in the head
Recommended by Juan Arroyo based on reviews by 3 anonymous reviewers
Plants display an amazing diversity of reproductive strategies with and without sex. This diversity is particularly remarkable in flowering plants, as highlighted by Charles Darwin, who wrote several botanical books scrutinizing plant reproduction. One particularly influential work concerned floral variation [1]. Darwin recognized that flowers may present different forms within a single population, with or without sex specialization. The number of species concerned is small, but they display recurrent patterns, which made it possible for Darwin to invoke natural and sexual selection to explain them. Most of early evolutionary theory on the evolution of reproductive strategies was developed in the first half of the 20th century and was based on animals. However, the pioneering work by David Lloyd from the 1970s onwards excited interest in the diversity of plant sexual strategies as models for testing adaptive hypotheses and predicting reproductive outcomes [2]. The sex specialization of individual flowers and plants has since become one of the favorite topics of evolutionary biologists. However, attention has focused mostly on cases related to sex differentiation (dioecy and associated conditions [3]). Separate unisexual flower types on the same plant (monoecy and related cases, rendering the plant functionally hermaphroditic) have been much less studied, apart from their possible role in the evolution of dioecy [4] or their association with particular modes of pollination [5].
Two specific non-mutually exclusive hypotheses on the evolution of separate sexes in flowers (dicliny) have been proposed, both anchored in Lloyd’s views and Darwin’s legacy, with selfing avoidance and optimal limited resource allocation. Intermediate sex separation, in which sex morphs have different combinations of unisexual and hermaphrodite flowers, has been crucial for testing these hypotheses through comparative analyses of optimal conditions in suggested transitions. Again, cases in which floral unisexuality does not lead to sex separation have been studied much less than dioecious plants, at both the microevolutionary and macroevolutionary levels. It is surprising that the increasing availability of plant phylogenies and powerful methods for testing evolutionary transitions and correlations have not led to more studies, even though the frequency of monoecy is probably highest among diclinous species (those with unisexual flowers in any distribution among plants within a population [6]).
The study by Torices et al. [7] aims to fill this gap, offering a different perspective to that provided by Diggle & Miller [8] on the evolution of monoecious conditions. The authors use heads of a number of species of the sunflower family (Asteraceae) to test specifically the effect of resource limitation on the expression of sexual morphs within the head. They make use of the very particular and constant architecture of inflorescences in these species (the flower head or “capitulum”) and the diversity of sexual conditions (hermaphrodite, gynomonoecious, monoecious) and their spatial pattern within the flower head in this plant family to develop an elegant means of testing this hypothesis. Their results are consistent with their expectations on the effect of resource limitation on the head, as determined by patterns of fruit size within the head, assuming that female fecundity is more strongly limited by resource availability than male function.
The authors took on a huge challenge in choosing to study the largest plant family (about 25 thousand species). Their sample was limited to only about a hundred species, but species selection was very careful, to ensure that the range of sex conditions and the available phylogenetic information were adequately represented. The analytical methods are robust and cast no doubt on the reported results. However, I can’t help but wonder what would happen if the antiselfing hypothesis was tested simultaneously. This would require self-incompatibility (SI) data for the species sample, as the presence of SI is usually invoked as a powerful antiselfing mechanism, rendering the unisexuality of flowers unnecessary. However, SI is variable and frequently lost in the sunflower family [9]. I also wonder to what extent the very specific architecture of flower heads imposes an idiosyncratic resource distribution that may have fixed these sexual systems in species and lineages of the family. Although not approached in this study, intraspecific variation seems to be low. It would be very interesting to use similar approaches in other plant groups in which inflorescence architecture is lax and resource distribution may differ. A whole-plant approach might be required, rather than investigations of single inflorescences as in this study. This study has no flaws, but instead paves the way for further testing of a long-standing dual hypothesis, probably with different outcomes in different ecological and evolutionary settings. In the end, sex is not only in the head.
References
[1] Darwin, C. (1877). The different forms of flowers on plants of the same species. John Murray.
[2] Barrett, S. C. H., and Harder, L. D. (2006). David G. Lloyd and the evolution of floral biology: from natural history to strategic analysis. In L.D. Harder, L. D., and Barrett, S. C. H. (eds) Ecology and Evolution of Flowers. OUP, Oxford. Pp 1-21.
[3] Geber, M. A., Dawson, T. E., and Delph, L. F. (eds) (1999). Gender and sexual dimorphism in flowering plants. Springer, Berlin.
[4] Charlesworth, D. (1999). Theories of the evolution of dioecy. In Geber, M. A., Dawson T. E. and Delph L. F. (eds) (1999). Gender and sexual dimorphism in flowering plants. Springer, Berlin. Pp. 33-60.
[5] Friedman, J., and Barrett, S. C. (2008). A phylogenetic analysis of the evolution of wind pollination in the angiosperms. International Journal of Plant Sciences, 169(1), 49-58. doi: 10.1086/523365
[6] Renner, S. S. (2014). The relative and absolute frequencies of angiosperm sexual systems: dioecy, monoecy, gynodioecy, and an updated online database. American Journal of botany, 101(10), 1588-1596. doi: 10.3732/ajb.1400196
[7] Torices, R., Afonso, A., Anderberg, A. A., Gómez, J. M., and Méndez, M. (2019). Architectural traits constrain the evolution of unisexual flowers and sexual segregation within inflorescences: an interspecific approach. bioRxiv, 356147, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/356147
[8] Diggle, P. K., and Miller, J. S. (2013). Developmental plasticity, genetic assimilation, and the evolutionary diversification of sexual expression in Solanum. American journal of botany, 100(6), 1050-1060. doi: 10.3732/ajb.1200647
[9] Ferrer, M. M., and Good‐Avila, S. V. (2007). Macrophylogenetic analyses of the gain and loss of self‐incompatibility in the Asteraceae. New Phytologist, 173(2), 401-414. doi: 10.1111/j.1469-8137.2006.01905.x
| Architectural traits constrain the evolution of unisexual flowers and sexual segregation within inflorescences: an interspecific approach | Rubén Torices, Ana Afonso, Arne A. Anderberg, José M. Gómez and Marcos Méndez | <p>Male and female unisexual flowers have repeatedly evolved from the ancestral bisexual flowers in different lineages of flowering plants. This sex specialization in different flowers often occurs within inflorescences. We hypothesize that inflor... |  | Evolutionary Ecology, Morphological Evolution, Phenotypic Plasticity, Reproduction and Sex, Sexual Selection | Juan Arroyo | Jana Vamosi, Marcial Escudero, Anonymous | 2018-06-27 10:49:52 | View |
Genome plasticity in Papillomaviruses and de novo emergence of E5 oncogenes
Anouk Willemsen, Marta Félez-Sánchez, and Ignacio G. Bravo
https://doi.org/10.1101/337477
E5, the third oncogene of Papillomavirus
Recommended by Hirohisa Kishino based on reviews by Leonardo de Oliveira Martins and 1 anonymous reviewer
Papillomaviruses (PVs) infect almost all mammals and possibly amniotes and bony fishes. While most of them have no significant effects on the hosts, some induce physical lesions. Phylogeny of PVs consists of a few crown groups [1], among which AlphaPVs that infect primates including human have been well studied. They are associated to largely different clinical manifestations: non-oncogenic PVs causing anogenital warts, oncogenic and non-oncogenic PVs causing mucosal lesions, and non-oncogenic PVs causing cutaneous warts.
The PV genome consists of a double stranded circular DNA genome, roughly organized into three parts: an early region coding for six open reading frames (ORFs: E1, E2, E4, E5, E6 and E7) involved in multiple functions including viral replication and cell transformation; a late region coding for structural proteins (L1 and L2); and a non-coding regulatory region (URR) that contains the cis-elements necessary for replication and transcription of the viral genome.
The E5, E6, and E7 are known to act as oncogenes. The E6 protein binds to the cellular p53 protein [2]. The E7 protein binds to the retinoblastoma tumor suppressor gene product, pRB [3]. However, the E5 has been poorly studied, even though a high correlation between the type of E5 protein and the infection phenotype is observed. E5s, being present on the E2/L2 intergenic region in the genomes of a few polyphyletic PV lineages, are so diverged and can only be characterized by high hydrophobicity. No similar sequences have been found in the sequence database.
Willemsen et al. [4] provide valuable evidence on the origin and evolutionary history of E5 genes and their genomic environments. First, they tested common ancestry vs independent origins [5]. Because alignment can lead to biased testing toward the hypothesis of common ancestry [6], they took full account of alignment uncertainty [7] and conducted random permutation test [8]. Although the strong chemical similarity hampered decisive conclusion on the test, they could confirm that E5 may do code proteins, and have unique evolutionary history with far different topology from the neighboring genes.
Still, there is mysteries with the origin and evolution of E5 genes. One of the largest interest may be the evolution of hydrophobicity, because it may be the main cause of variable infection phenotype. The inference has some similarity in nature with the inference of evolutionary history of G+C contents in bacterial genomes [9]. The inference may take account of possible opportunity of convergent or parallel evolution by setting an anchor to the topologies of neighboring genes.
References
[1] Bravo, I. G., & Alonso, Á. (2004). Mucosal human papillomaviruses encode four different E5 proteins whose chemistry and phylogeny correlate with malignant or benign growth. Journal of virology, 78, 13613-13626. doi: 10.1128/JVI.78.24.13613-13626.2004
[2] Werness, B. A., Levine, A. J., & Howley, P. M. (1990). Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science, 248, 76-79. doi: 10.1126/science.2157286
[3] Dyson, N., Howley, P. M., Munger, K., & Harlow, E. D. (1989). The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science, 243, 934-937. doi: 10.1126/science.2537532
[4] Willemsen, A., Félez-Sánchez, M., & Bravo, I. G. (2019). Genome plasticity in Papillomaviruses and de novo emergence of E5 oncogenes. bioRxiv, 337477, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/337477
[5] Theobald, D. L. (2010). A formal test of the theory of universal common ancestry. Nature, 465, 219–222. doi: 10.1038/nature09014
[6] Yonezawa, T., & Hasegawa, M. (2010). Was the universal common ancestry proved?. Nature, 468, E9. doi: 10.1038/nature09482
[7] Redelings, B. D., & Suchard, M. A. (2005). Joint Bayesian estimation of alignment and phylogeny. Systematic biology, 54(3), 401-418. doi: 10.1080/10635150590947041
[8] de Oliveira Martins, L., & Posada, D. (2014). Testing for universal common ancestry. Systematic biology, 63(5), 838-842. doi: 10.1093/sysbio/syu041
[9] Galtier, N., & Gouy, M. (1998). Inferring pattern and process: maximum-likelihood implementation of a nonhomogeneous model of DNA sequence evolution for phylogenetic analysis. Molecular biology and evolution, 15(7), 871-879. doi: 10.1093/oxfordjournals.molbev.a025991
| Genome plasticity in Papillomaviruses and de novo emergence of E5 oncogenes | Anouk Willemsen, Marta Félez-Sánchez, and Ignacio G. Bravo | <p>The clinical presentations of papillomavirus (PV) infections come in many different flavors. While most PVs are part of a healthy skin microbiota and are not associated to physical lesions, other PVs cause benign lesions, and only a handful of ... | 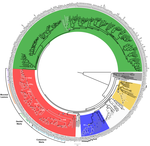 | Genome Evolution, Molecular Evolution, Phylogenetics / Phylogenomics | Hirohisa Kishino | | 2018-06-04 16:15:39 | View |
Replication-independent mutations: a universal signature ?
Recommended by Nicolas Galtier based on reviews by Marc Robinson-Rechavi and Robert Lanfear
Mutations are the primary source of genetic variation, and there is an obvious interest in characterizing and understanding the processes by which they appear. One particularly important question is the relative abundance, and nature, of replication-dependent and replication-independent mutations - the former arise as cells replicate due to DNA polymerization errors, whereas the latter are unrelated to the cell cycle. A recent experimental study in fission yeast identified a signature of mutations in quiescent (=non-replicating) cells: the spectrum of such mutations is characterized by an enrichment in insertions and deletions (indels) compared to point mutations, and an enrichment of deletions compared to insertions [2].
What Achaz et al. [1] report here is that the very same signature is detectable in humans. This time the approach is indirect and relies on two key aspects of mammalian reproduction biology: (1) oocytes remain quiescent over most of a female's lifespan, whereas spermatocytes keep dividing after male puberty, and (2) X chromosome, Y chromosome and autosomes spend different amounts of time in a female vs. male context. In agreement with the yeast study, Achaz et al. show that in humans the male-associated Y chromosome, for which quiescence is minimal, has by far the lowest ratios of indels to point mutations and of deletions to insertions, whereas the female-associated X chromosome has the highest. This is true both of variants that are polymorphic among humans and of fixed differences between humans and chimpanzees.
So we appear to be here learning about an important and general aspect of the mutation process. The authors suggest that, to a large extent, chromosomes tend to break in pieces at a rate that is proportional to absolute time - because indels in quiescent stage presumably result from double-strand DNA breaks. A very recent analysis of numerous mother-father-child trios in humans confirms this prediction in demonstrating an effect of maternal age, but not of paternal age, on the recombination rate [3]. This result also has important implications with respect to the interpretation of substitution rate variation among taxa and genomic compartments, particularly mitochondrial vs. nuclear, and their relationship with the generation time and longevity of organisms (e.g. [4]).
References
[1] Achaz, G., Gangloff, S., and Arcangioli, B. (2019). The quiescent X, the replicative Y and the Autosomes. BioRxiv, 351288, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/351288
[2] Gangloff, S., Achaz, G., Francesconi, S., Villain, A., Miled, S., Denis, C., and Arcangioli, B. (2017). Quiescence unveils a novel mutational force in fission yeast. eLife, 6:e27469. doi: 10.7554/eLife.27469
[3] Halldorsson, B. V., Palsson, G., Stefansson, O. A., Jonsson, H., Hardarson, M. T., Eggertsson, H. P., … Stefansson, K. (2019). Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science, 363: eaau1043. doi: 10.1126/science.aau1043
[4] Saclier, N., François, C. M., Konecny-Dupré, L., Lartillot, N., Guéguen, L., Duret, L., … Lefébure, T. (2019). Life History Traits Impact the Nuclear Rate of Substitution but Not the Mitochondrial Rate in Isopods. Molecular Biology and Evolution, in press. doi: 10.1093/molbev/msy247
| The quiescent X, the replicative Y and the Autosomes | Guillaume Achaz, Serge Gangloff, Benoit Arcangioli | <p>From the analysis of the mutation spectrum in the 2,504 sequenced human genomes from the 1000 genomes project (phase 3), we show that sexual chromosomes (X and Y) exhibit a different proportion of indel mutations than autosomes (A), ranking the... | 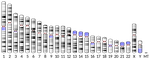 | Bioinformatics & Computational Biology, Genome Evolution, Human Evolution, Molecular Evolution, Population Genetics / Genomics, Reproduction and Sex | Nicolas Galtier | | 2018-07-25 10:37:48 | View |
A nice twist on partner choice theory
Recommended by Erol Akcay based on reviews by 2 anonymous reviewers
In this paper, Geoffroy et al. [1] deal with partner choice as a mechanism of maintaining cooperation, and argues that rather than being unequivocally a force towards improved payoffs to everyone through cooperation, partner choice can lead to “over-cooperation” where individuals can evolve to invest so much in cooperation that the costs of cooperating partially or fully negate the benefits from it. This happens when partner choice is consequential and effective, i.e., when interactions are long (so each decision to accept or reject a partner is a bigger stake) and when meeting new partners is frequent when unpaired (so that when one leaves an interaction one can find a new partner quickly). Geoffroy et al. [1] show that this tendency to select for overcooperation under such regimes can be counteracted if individuals base their acceptance-rejection of partners not just on the partner cooperativeness, but also on their own. By using tools from matching theory in economics, they show that plastic partner choice generates positive assortment between cooperativeness of the partners, and in the extreme case of perfectly assortative pairings, makes the pair the unit of selection, which selects for maximum total payoff.
This study is a nice contribution to the literature that illustrates potential complexities with partner choice as a mechanism for cooperation, including how the proximate mechanisms of partner choice can significantly alter the evolutionary trajectory of cooperation. Modeling choice as a reaction norm that depends on one’s own traits also adds a layer of realism to partner choice theory.
The authors are also to be commended for the revisions they made through the review process. Earlier versions of the model somewhat overstated the tendency for fixed partner choice strategies to lead to over cooperation, missing some of the important features in previous models, notably McNamara et al. [2] that can counter this tendency. In this version, the authors acknowledge these factors, mainly, mortality during partner choice (which increases the opportunity cost of forgoing a current partner) and also the fact that endogenous distribution of alternative partners (which will tend to be worse than the overall population distribution, because more cooperative types spend more time attached and less cooperative types more time unattached). These two factors can constrain cooperation from “running away” as the authors put it, but the main point of Geoffroy et al. [1] that plastic choice can create selection against inefficient cooperation stands.
I think the paper will be very stimulating to theoretical and empirical researchers working on partner choice and social behaviors, and happy to recommend it.
References
[1] Geoffroy, F., Baumard, N., & Andre, J.-B. (2019). Why cooperation is not running away. bioRxiv, ver. 5 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/316117
[2] McNamara, J. M., Barta, Z., Fromhage, L., & Houston, A. I. (2008). The coevolution of choosiness and cooperation. Nature, 451, 189–192. doi: 10.1038/nature06455
| Why cooperation is not running away | Félix Geoffroy, Nicolas Baumard, Jean-Baptiste André | <p>A growing number of experimental and theoretical studies show the importance of partner choice as a mechanism to promote the evolution of cooperation, especially in humans. In this paper, we focus on the question of the precise quantitative lev... | 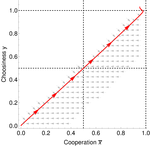 | Behavior & Social Evolution, Evolutionary Theory | Erol Akcay | | 2018-05-15 10:32:51 | View |





