The cell-level perspective in social conflicts in Dictyostelium discoideum
Recommended by Jeremy Van Cleve based on reviews by Peter Conlin and ?
The social amoeba Dictyostelium discoideum is an important model system for the study of cooperation and multicellularity as is has both unicellular and aggregative life phases. In the aggregative phase, which typically occurs when nutrients are limiting, individual cells eventually gather together to form a fruiting bodies whose spores may be dispersed to another, better, location and whose stalk cells, which support the spores, die. This extreme form of cooperation has been the focus of numerous studies that have revealed the importance genetic relatedness and kin selection (Hamilton 1964; Lehmann and Rousset 2014) in explaining the maintenance of this cooperative collective behavior (Strassmann et al. 2000; Kuzdzal-Fick et al. 2011; Strassmann and Queller 2011). However, much remains unknown with respect to how the interactions between individual cells, their neighbors, and their environment produce cooperative behavior at the scale of whole groups or collectives. In this preprint, Forget et al. (2021) describe how the D. discoideum system is crucial in this respect because it allows these cellular-level interactions to be studied in a systematic and tractable manner.
Spore bias, which is the tendency of a particular genotype or strain to disproportionately migrate to the spore instead of the stalk, is often used to define which strains are "cheaters" (positive spore bias) and which are "cooperative" (negative spore bias). Forget et al. (2021) note that spore bias depends on a number of stochastic factors including external drivers such as variation in environmental (or nutrient) quality and internal drivers like cell-cycle phase at the time of starvation. Spore bias is also affected by the social environment where the fraction of cheater strains in a spore may be limited by the ability of the remaining stalk cells to support the spore. The social environment can also affect cells through their differential responsiveness to the chemical factors that induce differentiation into stalk cells; responsiveness is partly a function of nutrient quality (Thompson and Kay 2000), which in turn can be a function of cell density. Thus, Forget et al. (2021) highlight a number of mechanisms that could generate frequency-dependent selection that would lead to the stable maintenance of multiple strains with different spore biases; in other words, both cheater and cooperative strains might stably coexist due to these cellular-level interactions.
The cellular-level interactions that Forget et al. (2021) highlight are particularly important because they pose a challenge evolutionary theory: some evolutionary models of social and collective behavior neglect or simplify these interactions. For example, Forget et al. (2021) note that the developmental, behavior, and environmental timescales relevant for Dictyostelium fruiting body formation all overlap. Evolutionary analyses often assume some of these timescales, for example developmental and behavior, are separate in order to simplify the analysis of any interactions. Thus, new theoretical work that allows these timescales to overlap may shed light on how cellular-level interactions can produce environmental, physiological, and behavioral feedbacks that drive the evolution of cooperation and other collective behaviors.
References
Forget, M., Adiba, S. and De Monte, S.(2021) Social conflicts in *Dictyostelium discoideum *: a matter of scales. HAL, hal-03088868, ver. 2 peer-reviewed and recommended by PCI Evolutionary Biology. https://hal.archives-ouvertes.fr/hal-03088868/
Hamilton, W. D. (1964). The genetical evolution of social behaviour. II. Journal of theoretical biology, 7(1), 17-52. doi: https://doi.org/10.1016/0022-5193(64)90039-6
Kuzdzal-Fick, J. J., Fox, S. A., Strassmann, J. E., and Queller, D. C. (2011). High relatedness is necessary and sufficient to maintain multicellularity in Dictyostelium. Science, 334(6062), 1548-1551. doi: https://doi.org/10.1126/science.1213272
Lehmann, L., and Rousset, F. (2014). The genetical theory of social behaviour. Philosophical Transactions of the Royal Society B: Biological Sciences, 369(1642), 20130357. doi: https://doi.org/10.1098/rstb.2013.0357
Strassmann, J. E., and Queller, D. C. (2011). Evolution of cooperation and control of cheating in a social microbe. Proceedings of the National Academy of Sciences, 108(Supplement 2), 10855-10862. doi: https://doi.org/10.1073/pnas.1102451108
Strassmann, J. E., Zhu, Y., & Queller, D. C. (2000). Altruism and social cheating in the social amoeba Dictyostelium discoideum. Nature, 408(6815), 965-967. doi: https://doi.org/10.1038/35050087
Thompson, C. R., & Kay, R. R. (2000). Cell-fate choice in Dictyostelium: intrinsic biases modulate sensitivity to DIF signaling. Developmental biology, 227(1), 56-64. doi: https://doi.org/10.1006/dbio.2000.9877
| Social Conflicts in Dictyostelium discoideum : A Matter of Scales | Forget, Mathieu; Adiba, Sandrine; De Monte, Silvia | <p>The 'social amoeba' Dictyostelium discoideum, where aggregation of genetically heterogeneous cells produces functional collective structures, epitomizes social conflicts associated with multicellular organization. 'Cheater' populations that hav... | 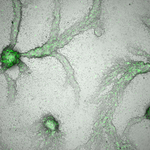 | Behavior & Social Evolution, Evolutionary Dynamics, Evolutionary Theory, Experimental Evolution | Jeremy Van Cleve | | 2020-08-28 10:37:21 | View |
Alteration of gut microbiota with a broad-spectrum antibiotic does not impair maternal care in the European earwig
Sophie Van Meyel, Séverine Devers, Simon Dupont, Franck Dedeine and Joël Meunier
https://doi.org/10.1101/2020.10.08.331363
Assessing the role of host-symbiont interactions in maternal care behaviour
Recommended by Trine Bilde based on reviews by Nadia Aubin-Horth, Gabrielle Davidson and 1 anonymous reviewer based on reviews by Nadia Aubin-Horth, Gabrielle Davidson and 1 anonymous reviewer
The role of microbial symbionts in governing social traits of their hosts is an exciting and developing research area. Just like symbionts influence host reproductive behaviour and can cause mating incompatibilities to promote symbiont transmission through host populations (Engelstadter and Hurst 2009; Correa and Ballard 2016; Johnson and Foster 2018) (see also discussion on conflict resolution in Johnsen and Foster 2018), microbial symbionts could enhance transmission by promoting the social behaviour of their hosts (Ezenwa et al. 2012; Lewin-Epstein et al. 2017; Gurevich et al. 2020). Here I apply the term ‘symbiosis’ in the broad sense, following De Bary 1879 as “the living together of two differently named organisms“ independent of effects on the organisms involved (De Bary 1879), i.e. the biological interaction between the host and its symbionts may include mutualism, parasitism and commensalism.
So far, we have relative few studies that explore the role of symbionts in promoting social behaviours such as parental care. Clearly, disentangling cause and effect when assessing the functional significance of symbiotic relationships in general is extremely challenging, and perhaps even more caution is needed when assessing the role of symbionts in the evolution of parental care, given the high fitness benefits to the offspring of receiving care. An interesting study on the symbiotic relationship between termites and their eukaryotic gut symbionts proposes a role of gut flagellates in the origin of subsocial behaviour (extended offspring care) in the termites through proctodeal trophallaxis (i.e. anus-to-mouth feeding), driven by mutualistic beneficial interactions (Nalepa 2020). Van Meyel et al. (2021) hypothesized a role of gut symbionts in promoting maternal care behaviour in the European earwig, and set out to test this idea in a carefully executed experimental study. They used a broad-spectrum antibiotic treatment to alter gut microbiota in mothers and examined its effect on maternal care provisioning. While the antibiotic treatment altered the gut microbiome, no effect on pre- or post-hatching maternal care was detected. The authors also investigated a broad range of physiological and reproductive traits measured over a major part of a female’s lifetime, and detected no effect of microbiome alteration on these traits. The study therefore does not provide evidence for a direct role of the gut microbiome in shaping offspring care in this population of European earwigs.
Within populations, earwigs show inter-individual variation in the expression of maternal care (Meunier et al. 2012; Ratz et al. 2016), and there is evidence that genetic and environmental factors contribute to this this variation (Meunier and Kolliker 2012; Kramer et al. 2017). The study by Van Meyel et al. (2021) is the first to analyse microbiome composition of the European earwig, and they study host-symbiont associations in a single population. A next step could be to explore among population variation in the gut microbiome, to achieve a better understanding on host-microbiome variation and dynamics in wild populations. Depending on the nature of host-symbiont associations across populations, new perspectives on their functional significance may arise (Hird 2017; Johnson and Foster 2018). It is therefore too early to conclusively confirm or reject the role of microbial symbionts in the expression of parental care in this system.
References
Correa, C. C., and Ballard, J. W. O. (2016). Wolbachia associations with insects: winning or losing against a master manipulator. Frontiers in Ecology and Evolution, 3, 153. doi: https://doi.org/10.3389/fevo.2015.00153
De Bary, A. (1879). Die Erscheinung der Symbiose. Verlag von Karl J. Trubner, Strassburg.
Engelstädter, J., and Hurst, G. D. (2009). The ecology and evolution of microbes that manipulate host reproduction. Annual Review of Ecology, Evolution, and Systematics, 40, 127-149. doi: https://doi.org/10.1146/annurev.ecolsys.110308.120206
Ezenwa, V. O., Gerardo, N. M., Inouye, D. W., Medina, M., and Xavier, J. B. (2012). Animal behavior and the microbiome. Science, 338(6104), 198-199. doi: https://doi.org/10.1126/science.1227412
Gurevich, Y., Lewin-Epstein, O., and Hadany, L. (2020). The evolution of paternal care: a role for microbes?. Philosophical Transactions of the Royal Society B, 375(1808), 20190599. doi: https://doi.org/10.1098/rstb.2019.0599
Hird, S. M. (2017). Evolutionary biology needs wild microbiomes. Frontiers in microbiology, 8, 725. doi: https://doi.org/10.3389/fmicb.2017.00725
Johnson, K. V. A., and Foster, K. R. (2018). Why does the microbiome affect behaviour?. Nature reviews microbiology, 16(10), 647-655. doi: https://doi.org/10.1038/s41579-018-0014-3
Kramer et al. (2017). When earwig mothers do not care to share: parent–offspring competition and the evolution of family life. Functional Ecology, 31(11), 2098-2107. doi: https://doi.org/10.1111/1365-2435.12915
Lewin-Epstein, O., Aharonov, R., and Hadany, L. (2017). Microbes can help explain the evolution of host altruism. Nature communications, 8(1), 1-7. doi: https://doi.org/10.1038/ncomms14040
Meunier, J., and Kölliker, M. (2012). Parental antagonism and parent–offspring co-adaptation interact to shape family life. Proceedings of the Royal Society B: Biological Sciences, 279(1744), 3981-3988. doi: https://doi.org/10.1098/rspb.2012.1416
Meunier, J., Wong, J. W., Gómez, Y., Kuttler, S., Röllin, L., Stucki, D., and Kölliker, M. (2012). One clutch or two clutches? Fitness correlates of coexisting alternative female life-histories in the European earwig. Evolutionary Ecology, 26(3), 669-682. doi: https://doi.org/10.1007/s10682-011-9510-x
Nalepa, C. A. (2020). Origin of mutualism between termites and flagellated gut protists: transition from horizontal to vertical transmission. Frontiers in Ecology and Evolution, 8, 14. doi: https://doi.org/10.3389/fevo.2020.00014
Ratz, T., Kramer, J., Veuille, M., and Meunier, J. (2016). The population determines whether and how life-history traits vary between reproductive events in an insect with maternal care. Oecologia, 182(2), 443-452. doi: https://doi.org/10.1007/s00442-016-3685-3
Van Meyel, S., Devers, S., Dupont, S., Dedeine, F. and Meunier, J. (2021) Alteration of gut microbiota with a broad-spectrum antibiotic does not impair maternal care in the European earwig. bioRxiv, 2020.10.08.331363. ver. 5 peer-reviewed and recommended by PCI Evol Biol. https://doi.org/10.1101/2020.10.08.331363
| Alteration of gut microbiota with a broad-spectrum antibiotic does not impair maternal care in the European earwig | Sophie Van Meyel, Séverine Devers, Simon Dupont, Franck Dedeine and Joël Meunier | <p>The microbes residing within the gut of an animal host often increase their own fitness by modifying their host’s physiological, reproductive, and behavioural functions. Whereas recent studies suggest that they may also shape host sociality and... |  | Behavior & Social Evolution, Evolutionary Ecology, Experimental Evolution, Life History, Species interactions | Trine Bilde | | 2020-10-09 14:07:47 | View |
Multi-gene and lineage comparative assessment of the strength of selection in Hymenoptera
Recommended by Bertanne Visser based on reviews by Michael Lattorff and 1 anonymous reviewer
Genetic variation is the raw material for selection to act upon and the amount of genetic variation present within a population is a pivotal determinant of a population’s evolutionary potential. A large effective population size, i.e., the ideal number of individuals experiencing the same amount of genetic drift and inbreeding as an actual population, Ne (Wright 1931, Crow 1954), thus increases the probability of long-term survival of a population. However, natural populations, as opposed to theoretical ones, rarely adhere to the requirements of an ideal panmictic population (Sjödin et al. 2005). A range of circumstances can reduce Ne, including the structuring of populations (through space and time, as well as age and developmental stages) and inbreeding (Charlesworth 2009). In mammals, species with a larger body mass (as a proxy for lower Ne) were found to have a higher rate of nonsynonymous nucleotide substitutions (that alter the amino acid sequence of a protein), as well as radical amino acid substitutions (altering the physicochemical properties of a protein) (Popadin et al. 2007). In general, low effective population sizes increase the chance of mutation accumulation and drift, while reducing the strength of selection (Sjödin et al. 2005).
In this paper, Weyna and Romiguier (2021) set out to test if parasitism, body size, geographic range, and/or eusociality affect the strength of selection in Hymenoptera. Hymenoptera include the bees, wasps and ants and is an extraordinarily diverse order within the insects. It was recently estimated that Hymenoptera is the most speciose order of the animal kingdom (Forbes et al. 2018). Hymenoptera are further characterized by an impressive radiation of parasitic species, mainly parasitoids, that feed in or on a single host individual to complete their own development (Godfray 1994). All hymenopterans share the same sex determination system: haplo-diploidy, where unfertilized eggs are haploid males and fertilized eggs are diploid females. Compared to other animals, Hymenoptera further contain an impressive number of clades that evolved eusociality (Rehan and Toth 2015), in which societies show a clear division of labor for reproduction (i.e., castes) and cooperative brood care. Hymenopterans thus represent a diverse and interesting group of insects to investigate potential factors affecting strength of selection and Ne.
Using a previously published phylogenomic dataset containing 3256 genes and 169 hymenopteran species (Peters et al. 2017), Weyna and Romiguier (2021) estimated mean genomic dN/dS ratios (nonsynonymous to synonymous substitution rates) for each species and compared these values between parasitic and non-parasitic species, eusocial and solitary species, and in relation to body size, parasitoid-specific traits and geographic range, thought to affect the effective population size and strength of selection. The use of a large number of species, as well as several distinct traits is a clear asset of this study. The authors found no effect of body size, geographic range or parasitism (including a range of parasitoid-specific traits). There was an effect, however, of eusociality where dN/dS increased in three out of four eusocial lineages. Future studies including more independent evolutionary transitions to eusociality can lend further support that eusocial species indeed reduces the efficiency of selection. The most intriguing result was that for solitary and social bees, with high dN/dS ratios and a strong signature of relaxed selection (i.e., the elimination or reduction of a source of selection (Lahti et al. 2009). The authors suggest that the pollen-collecting behaviors of these species can constrain Ne, as pollen availability varies at both a spatial and temporal scale, requiring a large investment in foraging that may in turn limit reproductive output. It would be interesting to see if other pollen feeders, such as certain beetles, flies, butterflies and moths, as well as mites and spiders, experience relaxed selection as a consequence of the trade-off between energy investment in pollen foraging versus fecundity.
References
Charlesworth, B. (2009). Effective population size and patterns of molecular evolution and variation. Nature Reviews Genetics, 10(3), 195-205. doi: https://doi.org/10.1038/nrg2526
Crow, J. F. (1954) Statistics and Mathematics in Biology (eds Kempthorne, O., Bancroft, T. A., Gowen, J. W. & Lush, J. L.) 543–556 (Iowa State Univ. Press, Ames, Iowa)
Forbes, A. A., Bagley, R. K., Beer, M. A., Hippee, A. C., and Widmayer, H. A. (2018). Quantifying the unquantifiable: why Hymenoptera, not Coleoptera, is the most speciose animal order. BMC ecology, 18(1), 1-11. doi: https://doi.org/10.1186/s12898-018-0176-x
Godfray, H. C. J. (1994) Parasitoids: Behavioral and Evolutionary Ecology. Vol. 67, Princeton University Press, 1994. doi: https://doi.org/10.2307/j.ctvs32rmp
Lahti et al. (2009). Relaxed selection in the wild. Trends in ecology & evolution, 24(9), 487-496. doi: https://doi.org/10.1016/j.tree.2009.03.010
Peters et al. (2017). Evolutionary history of the Hymenoptera. Current Biology, 27(7), 1013-1018. doi: https://doi.org/10.1016/j.cub.2017.01.027
Popadin, K., Polishchuk, L. V., Mamirova, L., Knorre, D., and Gunbin, K. (2007). Accumulation of slightly deleterious mutations in mitochondrial protein-coding genes of large versus small mammals. Proceedings of the National Academy of Sciences, 104(33), 13390-13395. doi: https://doi.org/10.1073/pnas.0701256104
Rehan, S. M., and Toth, A. L. (2015). Climbing the social ladder: the molecular evolution of sociality. Trends in ecology & evolution, 30(7), 426-433. doi: https://doi.org/10.1016/j.tree.2015.05.004
Sjödin, P., Kaj, I., Krone, S., Lascoux, M., and Nordborg, M. (2005). On the meaning and existence of an effective population size. Genetics, 169(2), 1061-1070. doi: https://doi.org/10.1534/genetics.104.026799
Weyna, A., and Romiguier, J. (2021) Relaxation of purifying selection suggests low effective population size in eusocial Hymenoptera and solitary pollinating bees. bioRxiv, 2020.04.14.038893, ver. 5 peer-reviewed and recommended by PCI Evol Biol. doi: https://doi.org/10.1101/2020.04.14.038893
Wright, S. (1931). Evolution in Mendelian populations. Genetics, 16(2), 97-159.
| Relaxation of purifying selection suggests low effective population size in eusocial Hymenoptera and solitary pollinating bees | Arthur Weyna, Jonathan Romiguier | <p>With one of the highest number of parasitic, eusocial and pollinator species among all insect orders, Hymenoptera features a great diversity of lifestyles. At the population genetic level, such life-history strategies are expected to decrease e... | 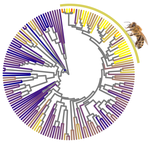 | Behavior & Social Evolution, Genome Evolution, Life History, Molecular Evolution, Population Genetics / Genomics | Bertanne Visser | | 2020-04-21 17:30:57 | View |
Another step towards grasping the complexity of the environmental response of traits
Recommended by Benoit Pujol based on reviews by 2 anonymous reviewers
One can only hope that one day, we will be able to evaluate how the ecological complexity surrounding natural populations affects their ability to adapt. This is more like a long term quest than a simple scientific aim. Many steps are heading in the right direction. This paper by Monroe and colleagues (2021) is one of them.
Many ecological and genetic mechanisms shape the evolutionary potential of phenotypic trait variation and many of them involve environmental heterogeneity (Pujol et al 2018). To date, we cannot look into these ecological and genetic mechanisms without oversimplifying their effects. We often look into trait variation one trait at a time albeit the variation of multiple phenotypic traits is often linked at the genetic or environmental level. As a consequence, we put our conclusions at risk by not accounting for the reciprocal impacts of trait changes upon each other (Teplitsky et al 2014). We also usually restrict the study of a continuous gradient of environmental conditions to a few conditions because it would otherwise be impossible to model its environmental effect. As a consequence, we miss the full picture of the continuous often nonlinear phenotypic plastic response. Whether the trait undergo threshold effect changes thereby remains obscured to us. Collectively, these issues impede our ability to understand how selection shapes the ecological strategy of organisms under variable environments.
In this paper, Monroe and colleagues (2021) propose an original approach that raised to these two challenges. They analysed phenotypic plastic changes in response to a continuous environment in a multidimensional trait space, namely the response of Brachypodium plant developmental and physiological traits to a continuous gradient of soil moisture. They used dry down experimental treatments to produce the continuous soil moisture gradient and compared the plant capacity to use water between annual B. distachyon and perennial B. sylvaticum. Their results revealed the best mathematical functions that model the nonlinear curvature of the continuous plastic response of Brachypodium plants. This work reinforces our view that nonlinear plastic responses can result in greater or lesser trait values at any stage of the environmental gradient that were unexpected on the basis of linear predictors (Gienapp and Brommer 2014). Their findings also imply that different threshold responses characterize different genotypes. These could otherwise have been missed by a classical approach. By shedding light on unforeseen interactions between traits that make their correlation vary along the nonlinear response, they were able to describe more accurately Brachypodium ecological strategies and the changes in evolutionary constraints along the soil moisture gradient.
Their empirical approach allows to test what environmental conditions maximises the opportunity for selection to shape trait variation. For example, it revealed unforeseen divergence in potentially adaptive mechanisms or life history strategies – and not just trait values – between annual and perennial species of Brachypodium. Behind every environmental variation of the constraints to the future evolutionary change of multiple traits, we can expect that the evolutionary history of the populations shaped their trait genetic correlations. Investigating the nonlinear signature of adaptive evolution across continuous environments will get us into uncharted territory.
Our ability to predict the adaptive potential of species is limited. With their approach of continuous environmental gradients beyond linearity, Monroe and collaborators (2021) improve our understanding of plant phenotypic responses and open a brand new range of exciting developments. As they mention: "the opportunity for scaling up" their approach is big. To illustrate this prospect, I can easily think of an example: the quantitative genetic random regression model. This model allows to use any degree of genetic relatedness in a wild population to estimate the genetic variation of phenotypic plastic reaction norms (Nussey et al 2007, Pujol and Galaud 2013). However, in this approach, only a few modalities of the environmental gradient are used to model nonlinear phenotypic plastic responses. From there, it is rather intuitive. Combining the best of these two approaches (continuity of genetic relatedness in the wild & continuity of environmental gradient in experiments) could open ground breaking new perspectives in research.
References
Gienapp P. & J.E. Brommer. 2014. Evolutionary dynamics in response to climate change. In: Charmentier A, Garant D, Kruuk LEB, editors. Quantitative genetics in the wild. Oxford: Oxford University Press, Oxford. pp. 254–273. doi: https://doi.org/10.1093/acprof:oso/9780199674237.003.0015
Monroe, J. G., Cai, H., and Des Marais, D. L. (2020). Trait plasticity and covariance along a continuous soil moisture gradient. bioRxiv, 2020.02.17.952853, ver. 5 peer-reviewed and recommended by PCI Evol Biol. doi: https://doi.org/10.1101/2020.02.17.952853
Pujol et al. (2018). The missing response to selection in the wild. Trends in ecology & evolution, 33(5), 337-346. doi: https://doi.org/10.1016/j.tree.2018.02.007
Pujol, B., and Galaud, J. P. (2013). A practical guide to quantifying the effect of genes underlying adaptation in a mixed genomics and evolutionary ecology approach. Botany Letters, 160(3-4), 197-204. doi: https://doi.org/10.1080/12538078.2013.799045
Nussey, D. H., Wilson, A. J., and Brommer, J. E. (2007). The evolutionary ecology of individual phenotypic plasticity in wild populations. Journal of evolutionary biology, 20(3), 831-844. doi: https://doi.org/10.1111/j.1420-9101.2007.01300.x
Teplitsky et al. (2014). Assessing multivariate constraints to evolution across ten long-term avian studies. PLoS One, 9(3), e90444. doi: https://doi.org/10.1371/journal.pone.0090444
| Trait plasticity and covariance along a continuous soil moisture gradient | J Grey Monroe, Haoran Cai, David L Des Marais | <p>Water availability is perhaps the greatest environmental determinant of plant yield and fitness. However, our understanding of plant-water relations is limited because it is primarily informed by experiments considering soil moisture variabilit... |  | Phenotypic Plasticity | Benoit Pujol | | 2020-02-20 16:34:40 | View |
Shifts from pulled to pushed range expansions caused by reduction of landscape connectivity
Maxime Dahirel, Aline Bertin, Marjorie Haond, Aurélie Blin, Eric Lombaert, Vincent Calcagno, Simon Fellous, Ludovic Mailleret, Thibaut Malausa, Elodie Vercken
https://doi.org/10.1101/2020.05.13.092775
The push and pull between theory and data in understanding the dynamics of invasion
Recommended by Ben Phillips based on reviews by Laura Naslund and 2 anonymous reviewers
Exciting times are afoot for those of us interested in the ecology and evolution of invasive populations. Recent years have seen evolutionary process woven firmly into our understanding of invasions (Miller et al. 2020). This integration has inspired a welter of empirical and theoretical work. We have moved from field observations and verbal models to replicate experiments and sophisticated mathematical models. Progress has been rapid, and we have seen science at its best; an intimate discussion between theory and data.
An area currently under very active development is our understanding of pushed invasions. Here a population spreads through space driven, not by dispersal and growth originating at the leading tip of the invasion, but by dispersal and growth originating deeper in the bulk of the population. These pushed invasions may be quite common – they result when per capita growth and dispersal rates are higher in the bulk of the wave than at the leading tip. They result from a range of well-known phenomena, including Allee effects and density-dependent dispersal (Gandhi et al. 2016; Bîrzu et al. 2019). Pushed invasions travel faster than we would expect given growth and dispersal rates on the leading tip, and they lose genetic diversity more slowly than classical pulled invasions (Roques et al. 2012; Haond et al. 2018; Bîrzu et al. 2019).
Well… in theory, anyway. The theory on pushed waves has momentarily streaked ahead of the empirical work, because empirical systems for studying pushed invasions are rare (though see Gandhi et al. 2016; Gandhi, Korolev, and Gore 2019). In this paper, Dahirel and colleagues (2020) make the argument that we may be able to generate pushed invasions in laboratory systems simply by reducing the connectedness of our experimental landscapes. If true, we might have a simple tool for turning many of our established experimental systems into systems for studying pushed dynamics.
It’s a nice idea, and the paper goes to careful lengths to explore the possibility in their lab system (a parasitoid wasp, Trichogramma). They run experiments on replicate wasp populations comparing strongly- v poorly-connected arrays, and estimate the resulting invasion speeds and rate of diversity loss. They also build a simulation model of the system, allowing them to explore in-silico a range of possible processes underlying their results.
As well as developing these parallel systems, Dahirel and colleagues (2020) go to careful lengths to develop statistical analyses that allow inference on key parameters, and they apply these analyses to both the experimental and simulation data. They have been motivated to apply methods that might be used in both laboratory and field settings to help classify invasions.
Ultimately, they found reasonable evidence that their poorly-connected habitat did induce a pushed dynamic. Their poorly connected invasions travelled faster than they should have if they were pulled, they lost diversity more slowly than the highly connected habitat, and replicates with a higher carrying capacity tended to have higher invasion speeds. All in line with expectations of a pushed dynamic. Interestingly, however, their simulation results suggest that they probably got this perfect result for unexpected reasons. The strong hint is that their poorly-connected habitat induced density dependent dispersal in the wasps. Without this effect, their simulations suggest they should have seen diversity decreasing much more rapidly than it did.
There is a nuanced, thoughtful, and carefully argued discussion about all this in the paper, and it is worth reading. There is much of value in this paper. Theirs is not a perfect empirical system in which all the model assumptions are met and in which huge population sizes make stochastic effects negligible. Here is a system one step closer to the messy reality of biology. The struggle to align this system with new theory has been worth the effort. Not only does it give us hope that we might usefully be able to discriminate between classes of invasions using real-world data, but it hints at a rule that Tolstoy might have expressed this way: all pulled invasions are alike, each pushed invasion is pushed in its own way.
References
Bîrzu, G., Matin, S., Hallatschek, O., and Korolev, K. S. (2019). Genetic drift in range expansions is very sensitive to density dependence in dispersal and growth. Ecology Letters, 22(11), 1817-1827. doi: https://doi.org/10.1111/ele.13364
Dahirel, M., Bertin, A., Haond, M., Blin, A., Lombaert, E., Calcagno, V., Fellous, S., Mailleret, L., Malausa, T., and Vercken, E. (2020). Shifts from pulled to pushed range expansions caused by reduction of landscape connectivity. bioRxiv, 2020.05.13.092775, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. https://doi.org/10.1101/2020.05.13.092775
Gandhi, S. R., Korolev, K. S., and Gore, J. (2019). Cooperation mitigates diversity loss in a spatially expanding microbial population. Proceedings of the National Academy of Sciences, 116(47), 23582-23587. doi: https://doi.org/10.1073/pnas.1910075116
Gandhi, S. R., Yurtsev, E. A., Korolev, K. S., and Gore, J. (2016). Range expansions transition from pulled to pushed waves as growth becomes more cooperative in an experimental microbial population. Proceedings of the National Academy of Sciences, 113(25), 6922-6927. doi: https://doi.org/10.1073/pnas.1521056113
Haond, M., Morel-Journel, T., Lombaert, E., Vercken, E., Mailleret, L. and Roques, L. (2018). When higher carrying capacities lead to faster propagation (2018), bioRxiv, 307322, ver. 4 peer-reviewed and recommended by Peer Community in Ecology. https://doi.org/10.1101/307322
Miller et al. (2020). Eco‐evolutionary dynamics of range expansion. Ecology, 101(10), e03139. doi: https://doi.org/10.1002/ecy.3139
Roques, L., Garnier, J., Hamel, F., and Klein, E. K. (2012). Allee effect promotes diversity in traveling waves of colonization. Proceedings of the National Academy of Sciences, 109(23), 8828-8833. doi: https://doi.org/10.1073/pnas.1201695109
| Shifts from pulled to pushed range expansions caused by reduction of landscape connectivity | Maxime Dahirel, Aline Bertin, Marjorie Haond, Aurélie Blin, Eric Lombaert, Vincent Calcagno, Simon Fellous, Ludovic Mailleret, Thibaut Malausa, Elodie Vercken | <p>Range expansions are key processes shaping the distribution of species; their ecological and evolutionary dynamics have become especially relevant today, as human influence reshapes ecosystems worldwide. Many attempts to explain and predict ran... |  | Evolutionary Applications, Evolutionary Dynamics, Evolutionary Ecology, Experimental Evolution, Phylogeography & Biogeography | Ben Phillips | | 2020-08-04 12:51:56 | View |
Quantifying transmission dynamics of acute hepatitis C virus infections in a heterogeneous population using sequence data
Gonche Danesh, Victor Virlogeux, Christophe Ramière, Caroline Charre, Laurent Cotte, Samuel Alizon
https://doi.org/10.1101/689158
Phylodynamics of hepatitis C virus reveals transmission dynamics within and between risk groups in Lyon
Recommended by David Rasmussen based on reviews by Chris Wymant and Louis DuPlessis
Genomic epidemiology seeks to better understand the transmission dynamics of infectious pathogens using molecular sequence data. Phylodynamic methods have given genomic epidemiology new power to track the transmission dynamics of pathogens by combining phylogenetic analyses with epidemiological modeling. In recent year, applications of phylodynamics to chronic viral infections such as HIV and hepatitis C virus (HVC) have provided some of the best examples of how phylodynamic inference can provide valuable insights into transmission dynamics within and between different subpopulations or risk groups, allowing for more targeted interventions.
However, conducting phylodynamic inference under complex epidemiological models comes with many challenges. In some cases, it is not always straightforward or even possible to perform likelihood-based inference. Structured SIR-type models where infected individuals can belong to different subpopulations provide a classic example. In this case, the model is both nonlinear and has a high-dimensional state space due to tracking different types of hosts. Computing the likelihood of a phylogeny under such a model involves complex numerical integration or data augmentation methods [1]. In these situations, Approximate Bayesian Computation (ABC) provides an attractive alternative, as Bayesian inference can be performed without computing likelihoods as long as one can efficiently simulate data under the model to compare against empirical observations [2].
Previous work has shown how ABC approaches can be applied to fit epidemiological models to phylogenies [3,4]. Danesh et al. [5] further demonstrate the real world merits of ABC by fitting a structured SIR model to HCV data from Lyon, France. Using this model, they infer viral transmission dynamics between “classical” hosts (typically injected drug users) and “new” hosts (typically young MSM) and show that a recent increase in HCV incidence in Lyon is due to considerably higher transmission rates among “new” hosts . This study provides another great example of how phylodynamic analysis can help epidemiologists understand transmission patterns within and between different risk groups and the merits of expanding our toolkit of statistical methods for phylodynamic inference.
References
[1] Rasmussen, D. A., Volz, E. M., and Koelle, K. (2014). Phylodynamic inference for structured epidemiological models. PLoS Comput Biol, 10(4), e1003570. doi: https://doi.org/10.1371/journal.pcbi.1003570
[2] Beaumont, M. A., Zhang, W., and Balding, D. J. (2002). Approximate Bayesian computation in population genetics. Genetics, 162(4), 2025-2035.
[3] Ratmann, O., Donker, G., Meijer, A., Fraser, C., and Koelle, K. (2012). Phylodynamic inference and model assessment with approximate bayesian computation: influenza as a case study. PLoS Comput Biol, 8(12), e1002835. doi: https://doi.org/10.1371/journal.pcbi.1002835
[4] Saulnier, E., Gascuel, O., and Alizon, S. (2017). Inferring epidemiological parameters from phylogenies using regression-ABC: A comparative study. PLoS computational biology, 13(3), e1005416. doi: https://doi.org/10.1371/journal.pcbi.1005416
[5] Danesh, G., Virlogeux, V., Ramière, C., Charre, C., Cotte, L. and Alizon, S. (2020) Quantifying transmission dynamics of acute hepatitis C virus infections in a heterogeneous population using sequence data. bioRxiv, 689158, ver. 5 peer-reviewed and recommended by PCI Evol Biol. doi: https://doi.org/10.1101/689158
| Quantifying transmission dynamics of acute hepatitis C virus infections in a heterogeneous population using sequence data | Gonche Danesh, Victor Virlogeux, Christophe Ramière, Caroline Charre, Laurent Cotte, Samuel Alizon | <p>Opioid substitution and syringes exchange programs have drastically reduced hepatitis C virus (HCV) spread in France but HCV sexual transmission in men having sex with men (MSM) has recently arisen as a significant public health concern. The fa... |  | Evolutionary Epidemiology, Phylogenetics / Phylogenomics | David Rasmussen | | 2019-07-11 13:37:23 | View |
Wolbachia and host intrinsic reproductive barriers contribute additively to post-mating isolation in spider mites
Miguel A. Cruz, Sara Magalhães, Élio Sucena, Flore Zélé
https://doi.org/10.1101/2020.06.29.178699
Speciation in spider mites: disentangling the roles of Wolbachia-induced vs. nuclear mating incompatibilities
Recommended by Jan Engelstaedter based on reviews by Wolfgang Miller and 1 anonymous reviewer
Cytoplasmic incompatibility (CI) is a mating incompatibility that is induced by maternally inherited endosymbionts in many arthropods. These endosymbionts include, most famously, the alpha-proteobacterium Wolbachia pipientis (Yen & Barr 1971; Werren et al. 2008) but also the Bacteroidetes bacterium Cardinium hertigii (Zchori-Fein et al. 2001), a gamma-proteobacterium of the genus Rickettsiella (Rosenwald et al. 2020) and another, as yet undescribed alpha-proteobacterium (Takano et al. 2017). CI manifests as embryonic mortality in crosses between infected males and females that are uninfected or infected with a different strain, whereas embryos develop normally in all other crosses. This phenotype may enable the endosymbionts to spread rapidly within their host population. Exploiting this, CI-inducing Wolbachia are being harnessed to control insect-borne diseases (e.g., O'Neill 2018). Much progress elucidating the genetic basis and developmental mechanism of CI has been made in recent years, but many open questions remain (Shropshire et al. 2020).
Immediately following the discovery and early study of CI in mosquitoes, Laven (1959, 1967) proposed that CI could be an important driver of speciation. Indeed, bi-directional CI can strongly reduce gene flow between two populations due to the elimination of F1 embryos, so that CI can act as a trigger for genetic differentiation in the host (Telschow et al. 2002, 2005). This idea has received much attention, and a potential role for CI in incipient speciation has been demonstrated in several species (e.g., Bordenstein et al. 2001; Jaenike et al. 2006). However, we still don’t know how commonly CI actually triggers speciation, rather than being merely a minor player or secondary phenomenon. The problem is that in addition to CI, postzygotic reproductive isolation can also be caused by host-induced, nuclear incompatibilities. Determining the relative contributions of these two causes of isolation is difficult and has rarely been done.
The study by Cruz et al. (2020) addresses this problem head-on, using a study system of Tetranychus urticae spider mites. These cosmopolitan mites are infected with different strains of Wolbachia. They come in two different colour forms (red and green) that can co-occur sympatrically on the same host plant but exhibit various degrees of reproductive isolation. A complicating factor in spider mites is that they are haplodiploid: unfertilised eggs develop into haploid males and are therefore not affected by any postzygotic incompatibilities, whereas fertilised eggs normally develop into diploid females. In haplodiploids, Wolbachia-induced CI can either kill diploid embryos (as in diplodiploid species), or turn them into haploid males. In their study, Cruz et al. used three different populations (one of the green and two of the red form) and employed a full factorial experiment involving all possible combinations of crosses of Wolbachia infected or uninfected males and females. For each cross, they measured F1 embryonic and juvenile mortality as well as sex ratio, and they also measured F1 fertility and F2 viability. Their results showed that there is strong reduction in hybrid female production caused by Wolbachia-induced CI. However, independent of this and through a different mechanism, there is an even stronger reduction in hybrid production caused by host-associated incompatibilities. In combination with the also observed near-complete sterility of F1 hybrid females and full F2 hybrid breakdown (neither of which is caused by Wolbachia), the results indicate essentially complete reproductive isolation between the green and red forms of T. urticae.
Overall, this is an elegant study with an admirably clean and comprehensive experimental design. It demonstrates that Wolbachia can contribute to reproductive isolation between populations, but that host-induced mechanisms of reproductive isolation predominate in these spider mite populations. Further studies in this exiting system would be useful that also investigate the contribution of pre-zygotic isolation mechanisms such as assortative mating, ascertain whether the results can be generalised to other populations, and – most challengingly – establish the order in which the different mechanisms of reproductive isolation evolved.
References
Bordenstein, S. R., O'Hara, F. P., and Werren, J. H. (2001). Wolbachia-induced incompatibility precedes other hybrid incompatibilities in Nasonia. Nature, 409(6821), 707-710. doi: https://doi.org/10.1038/35055543
Cruz, M. A., Magalhães, S., Sucena, É., and Zélé, F. (2020) Wolbachia and host intrinsic reproductive barriers contribute additively to post-mating isolation in spider mites. bioRxiv, 2020.06.29.178699, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: https://doi.org/10.1101/2020.06.29.178699
Jaenike, J., Dyer, K. A., Cornish, C., and Minhas, M. S. (2006). Asymmetrical reinforcement and Wolbachia infection in Drosophila. PLoS Biol, 4(10), e325. doi: https://doi.org/10.1371/journal.pbio.0040325
Laven, H. (1959). SPECIATION IN MOSQUITOES Speciation by Cytoplasmic Isolation in the Culex Pipiens-Complex. In Cold Spring Harbor Symposia on Quantitative Biology (Vol. 24, pp. 166-173). Cold Spring Harbor Laboratory Press.
Laven, H. (1967). A possible model for speciation by cytoplasmic isolation in the Culex pipiens complex. Bulletin of the World Health Organization, 37(2), 263-266.
O’Neill S.L. (2018) The Use of Wolbachia by the World Mosquito Program to Interrupt Transmission of Aedes aegypti Transmitted Viruses. In: Hilgenfeld R., Vasudevan S. (eds) Dengue and Zika: Control and Antiviral Treatment Strategies. Advances in Experimental Medicine and Biology, vol 1062. Springer, Singapore. doi: https://doi.org/10.1007/978-981-10-8727-1_24
Rosenwald, L.C., Sitvarin, M.I. and White, J.A. (2020). Endosymbiotic Rickettsiella causes cytoplasmic incompatibility in a spider host. doi: https://doi.org/10.1098/rspb.2020.1107
Shropshire, J. D., Leigh, B., and Bordenstein, S. R. (2020). Symbiont-mediated cytoplasmic incompatibility: what have we learned in 50 years?. Elife, 9, e61989. doi: https://doi.org/10.7554/eLife.61989
Takano et al. (2017). Unique clade of alphaproteobacterial endosymbionts induces complete cytoplasmic incompatibility in the coconut beetle. Proceedings of the National Academy of Sciences, 114(23), 6110-6115. doi: https://doi.org/10.1073/pnas.1618094114
Telschow, A., Hammerstein, P., and Werren, J. H. (2002). The effect of Wolbachia on genetic divergence between populations: models with two-way migration. the american naturalist, 160(S4), S54-S66. doi: https://doi.org/10.1086/342153
Telschow, A., Hammerstein, P., and Werren, J. H. (2005). The effect of Wolbachia versus genetic incompatibilities on reinforcement and speciation. Evolution, 59(8), 1607-1619. doi: https://doi.org/10.1111/j.0014-3820.2005.tb01812.x
Werren, J. H., Baldo, L., and Clark, M. E. (2008). Wolbachia: master manipulators of invertebrate biology. Nature Reviews Microbiology, 6(10), 741-751. doi: https://doi.org/10.1038/nrmicro1969
Yen, J. H., and Barr, A. R. (1971). New hypothesis of the cause of cytoplasmic incompatibility in Culex pipiens L. Nature, 232(5313), 657-658. doi: https://doi.org/10.1038/232657a0
Zchori-Fein, E., Gottlieb, Y., Kelly, S. E., Brown, J. K., Wilson, J. M., Karr, T. L., and Hunter, M. S. (2001). A newly discovered bacterium associated with parthenogenesis and a change in host selection behavior in parasitoid wasps. Proceedings of the National Academy of Sciences, 98(22), 12555-12560. doi: https://doi.org/10.1073/pnas.221467498
| Wolbachia and host intrinsic reproductive barriers contribute additively to post-mating isolation in spider mites | Miguel A. Cruz, Sara Magalhães, Élio Sucena, Flore Zélé | <p>Wolbachia are widespread maternally-inherited bacteria suggested to play a role in arthropod host speciation through induction of cytoplasmic incompatibility, but this hypothesis remains controversial. Most studies addressing Wolbachia-induced ... |  | Evolutionary Ecology, Hybridization / Introgression, Life History, Reproduction and Sex, Speciation, Species interactions | Jan Engelstaedter | | 2020-07-09 10:18:28 | View |
Sexual selection goes dynamic
Recommended by Michael D Greenfield based on reviews by Frédéric Guillaume and 1 anonymous reviewer
150 years after Darwin published ‘Descent of man and selection in relation to sex’ (Darwin, 1871), the evolutionary mechanism that he laid out in his treatise continues to fascinate us. Sexual selection is responsible for some of the most spectacular traits among animals, and plants, and it appeals to our interest in all things reproductive and sexual (Bell, 1982). In addition, sexual selection poses some of the more intractable problems in evolutionary biology: Its realm encompasses traits that are subject to markedly different selection pressures, particularly when distinct, yet associated, traits tend to be associated with males, e.g. courtship signals, and with females, e.g. preferences (cf. Ah-King & Ahnesjo, 2013). While separate, such traits cannot evolve independently of each other (Arnqvist & Rowe, 2005), and complex feedback loops and correlations between them are predicted (Greenfield et al., 2014). Traditionally, sexual selection has been modelled under simplifying assumptions, and quantitative genetic approaches that avoided evolutionary dynamics have prevailed. New computing methods may be able to free the field from these constraints, and a trio of theoreticians (Chevalier, De Coligny & Labonne 2020) describe here a novel application of a ‘demo-genetic agent (or individual) based model’, a mouthful hereafter termed DG-ABM, for arriving at a holistic picture of the sexual selection trajectory. The application is built on the premise that traits, e.g. courtship, preference, gamete investment, competitiveness for mates, can influence the genetic architecture, e.g. correlations, of those traits. In turn, the genetic architecture can influence the expression and evolvability of the traits. Much of this influence occurs via demographic features, i.e. social environment, generated by behavioral interactions during sexual advertisement, courtship, mate guarding, parental care, post-mating dispersal, etc.
The authors provide a lengthy verbal description of their model, specifying the genomic and behavioral parameters that can be set and how a ‘run’ may be initialized. There is a link to an internet site where users can then enter their own parameter values and begin exploring hypotheses. Back in the article several simulations illustrate simple tests; e.g. how gamete investment and preference jointly evolve given certain survival costs. One obvious test would have been the preference – courtship genetic correlation that represents the core of Fisherian runaway selection, and it is regrettable that it was not examined under a range of demographic parameters. As presented the author’s DG-ABM appears particularly geared toward mating systems in ‘higher’ vertebrates, where couples form during a discrete mating season and are responsible for most reproduction. It is not clear how applicable the model could be to a full range of mating systems and nuances, including those in arthropods and other invertebrates as well as plants.
What is the likely value of the DG-ABM for sexual selection researchers? We will not be able to evaluate its potential impact until readers with specialized understanding of a question and taxon begin exploring and comparing their results with prior expectations. Of course, lack of congruence with earlier predictions would not invalidate the model. Hopefully, some of these specialists will have opportunities for comparing results with pertinent empirical data.
References
Ah-King, M. and Ahnesjo, I. 2013. The ‘sex role’ concept: An overview and evaluation Evolutionary Biology, 40, 461-470. doi: https://doi.org/10.1007/s11692-013-9226-7
Arnqvist, G. and Rowe, L. 2005. Sexual Conflict. Princeton University Press, Princeton. doi: https://doi.org/10.1515/9781400850600
Bell, G. 1982. The Masterpiece of Nature: The Evolution and Genetics of Sexuality. University of California Press, Berkeley.
Chevalier, L., De Coligny, F. and Labonne, J. (2020) A demogenetic individual based model for the evolution of traits and genome architecture under sexual selection. bioRxiv, 2020.04.01.014514, ver. 4 peer-reviewed and recommended by PCI Evol Biol. doi: https://doi.org/10.1101/2020.04.01.014514
Darwin, C. 1871. The Descent of Man and Selection in Relation to Sex. J. Murray, London.
Greenfield, M.D., Alem, S., Limousin, D. and Bailey, N.W. 2014. The dilemma of Fisherian sexual selection: Mate choice for indirect benefits despite rarity and overall weakness of trait-preference genetic correlation. Evolution, 68, 3524-3536. doi: https://doi.org/10.1111/evo.12542
| A demogenetic agent based model for the evolution of traits and genome architecture under sexual selection | Louise Chevalier, François de Coligny, Jacques Labonne | <p>Sexual selection has long been known to favor the evolution of mating behaviors such as mate preference and competitiveness, and to affect their genetic architecture, for instance by favoring genetic correlation between some traits. Reciprocall... | 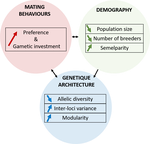 | Adaptation, Behavior & Social Evolution, Evolutionary Dynamics, Evolutionary Theory, Life History, Population Genetics / Genomics, Sexual Selection | Michael D Greenfield | | 2020-04-02 14:44:25 | View |
Review and Assessment of Performance of Genomic Inference Methods based on the Sequentially Markovian Coalescent
Recommended by Stephan Schiffels based on reviews by 3 anonymous reviewers
The human genome not only encodes for biological functions and for what makes us human, it also encodes the population history of our ancestors. Changes in past population sizes, for example, affect the distribution of times to the most recent common ancestor (tMRCA) of genomic segments, which in turn can be inferred by sophisticated modelling along the genome.
A key framework for such modelling of local tMRCA tracts along genomes is the Sequentially Markovian Coalescent (SMC) (McVean and Cardin 2005, Marjoram and Wall 2006) . The problem that the SMC solves is that the mosaic of local tMRCAs along the genome is unknown, both in their actual ages and in their positions along the genome. The SMC allows to effectively sum across all possibilities and handle the uncertainty probabilistically. Several important tools for inferring the demographic history of a population have been developed built on top of the SMC, including PSMC (Li and Durbin 2011), diCal (Sheehan et al 2013), MSMC (Schiffels and Durbin 2014), SMC++ (Terhorst et al 2017), eSMC (Sellinger et al. 2020) and others.
In this paper, Sellinger, Abu Awad and Tellier (2020) review these SMC-based methods and provide a coherent simulation design to comparatively assess their strengths and weaknesses in a variety of demographic scenarios (Sellinger, Abu Awad and Tellier 2020). In addition, they used these simulations to test how breaking various key assumptions in SMC methods affects estimates, such as constant recombination rates, or absence of false positive SNP calls.
As a result of this assessment, the authors not only provide practical guidance for researchers who want to use these methods, but also insights into how these methods work. For example, the paper carefully separates sources of error in these methods by observing what they call “Best-case convergence” of each method if the data behaves perfectly and separating that from how the method applies with actual data. This approach provides a deeper insight into the methods than what we could learn from application to genomic data alone.
In the age of genomics, computational tools and their development are key for researchers in this field. All the more important is it to provide the community with overviews, reviews and independent assessments of such tools. This is particularly important as sometimes the development of new methods lacks primary visibility due to relevant testing material being pushed to Supplementary Sections in papers due to space constraints. As SMC-based methods have become so widely used tools in genomics, I think the detailed assessment by Sellinger et al. (2020) is timely and relevant.
In conclusion, I recommend this paper because it bridges from a mere review of the different methods to an in-depth assessment of performance, thereby addressing both beginners in the field who just seek an initial overview, as well as experienced researchers who are interested in theoretical boundaries and assumptions of the different methods.
References
[1] Li, H., and Durbin, R. (2011). Inference of human population history from individual whole-genome sequences. Nature, 475(7357), 493-496. doi: https://doi.org/10.1038/nature10231
[2] Marjoram, P., and Wall, J. D. (2006). Fast"" coalescent"" simulation. BMC genetics, 7(1), 16. doi: https://doi.org/10.1186/1471-2156-7-16
[3] McVean, G. A., and Cardin, N. J. (2005). Approximating the coalescent with recombination. Philosophical Transactions of the Royal Society B: Biological Sciences, 360(1459), 1387-1393. doi: https://doi.org/10.1098/rstb.2005.1673
[4] Schiffels, S., and Durbin, R. (2014). Inferring human population size and separation history from multiple genome sequences. Nature genetics, 46(8), 919-925. doi: https://doi.org/10.1038/ng.3015
[5] Sellinger, T. P. P., Awad, D. A., Moest, M., and Tellier, A. (2020). Inference of past demography, dormancy and self-fertilization rates from whole genome sequence data. PLoS Genetics, 16(4), e1008698. doi: https://doi.org/10.1371/journal.pgen.1008698
[6] Sellinger, T. P. P., Awad, D. A. and Tellier, A. (2020) Limits and Convergence properties of the Sequentially Markovian Coalescent. bioRxiv, 2020.07.23.217091, ver. 3 peer-reviewed and recommended by PCI Evolutionary Biology. doi: https://doi.org/10.1101/2020.07.23.217091
[7] Sheehan, S., Harris, K., and Song, Y. S. (2013). Estimating variable effective population sizes from multiple genomes: a sequentially Markov conditional sampling distribution approach. Genetics, 194(3), 647-662. doi: https://doi.org/10.1534/genetics.112.149096
[8] Terhorst, J., Kamm, J. A., and Song, Y. S. (2017). Robust and scalable inference of population history from hundreds of unphased whole genomes. Nature genetics, 49(2), 303-309. doi: https://doi.org/10.1038/ng.3748
| Limits and Convergence properties of the Sequentially Markovian Coalescent | Thibaut Sellinger, Diala Abu Awad, Aurélien Tellier | <p>Many methods based on the Sequentially Markovian Coalescent (SMC) have been and are being developed. These methods make use of genome sequence data to uncover population demographic history. More recently, new methods have extended the original... | 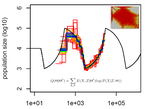 | Population Genetics / Genomics | Stephan Schiffels | Anonymous | 2020-07-25 10:54:48 | View |
A genomic amplification affecting a carboxylesterase gene cluster confers organophosphate resistance in the mosquito Aedes aegypti: from genomic characterization to high-throughput field detection
Julien Cattel, Chloé Haberkorn, Fréderic Laporte, Thierry Gaude, Tristan Cumer, Julien Renaud, Ian W. Sutherland, Jeffrey C. Hertz, Jean-Marc Bonneville, Victor Arnaud, Camille Noûs, Bénédicte Fustec, Sébastien Boyer, Sébastien Marcombe, Jean-Philippe David
https://doi.org/10.1101/2020.06.08.139741
Identification of a gene cluster amplification associated with organophosphate insecticide resistance: from the diversity of the resistance allele complex to an efficient field detection assay
Recommended by Stephanie Bedhomme based on reviews by Diego Ayala and 2 anonymous reviewers
The emergence and spread of insecticide resistance compromises the efficiency of insecticides as prevention tool against the transmission of insect-transmitted diseases (Moyes et al. 2017). In this context, the understanding of the genetic mechanisms of resistance and the way resistant alleles spread in insect populations is necessary and important to envision resistance management policies. A common and important mechanism of insecticide resistance is gene amplification and in particular amplification of insecticide detoxification genes, which leads to the overexpression of these genes (Bass & Field, 2011). Cattel and coauthors (2020) adopt a combination of experimental approaches to study the role of gene amplification in resistance to organophosphate insecticides in the mosquito Aedes aegypti and its occurrence in populations of South East Asia and to develop a molecular test to track resistance alleles.
Their first approach consists in performing an artificial selection on laboratory Ae. Aegypti populations started with individuals collected in Laos. In the selected population, an initial 90% mortality by adult exposure to the organophosphate insecticide malathion is imposed. This population shows a steep increase in resistance to malathion and other organophosphate insecticides, which is absent in the paired control population. The transcriptomic patterns of the control and the evolved populations as well as of a reference sensitive population reveals, among other differences, the over-expression of five carboxy/choline esterase (CCE) genes in the insecticide selected population. These five genes happen to be clustered in the Ae. aegypti genome and whole genome sequencing of a highly resistant population combined to qPCR test on genomic DNA showed that the overexpression of these genes is due to gene amplification. Although it would have been more elegant to have replicate selected and control populations and to perform the transcriptomic and the genomic analyses directly on the experimental populations, the authors gather a set of experimental evidence which combined to previous knowledge on the function of the amplified and over-expressed genes and on their implication in organophosphate insecticide resistance in other species allow to discard the possibility that this gene amplification spread by drift in the selected population.
In a second part of the paper, copy number variation for CCE genes is checked in field sample populations. This test reveals the presence of resistance alleles in half of the fourteen South East Asia populations sampled. Very interestingly, it also reveals a high level of complexity and diversity among the resistance alleles: it shows first the existence, both in the experimental and the field populations, of at least two amplified alleles (differing by the number of genes amplified) and second a high variation in the copy number of amplified genes. This indicates that gene amplification as a molecular resistance mechanism has actually lead to a high diversity of resistance alleles. These alleles are likely to differ both by the level of resistance conferred and the fitness cost imposed in the absence of the insecticide and these two values are affecting the evolution of their frequency in the field and ultimately the spread of resistance.
The last part of the paper is devoted to the development of a high-throughput Taqman assay which allows to determine rapidly the copy number of one of the esterase genes amplified in the resistance alleles described earlier. This assay is nicely validated and will definitely be a useful tool to determine the occurrence of these resistance alleles in field population. The fact that it gives access to the copy number will also allow to follow its copy number across time and get insight into the complexity of resistance evolution by gene amplification.
To sum up, this paper studies the implication of carboxy/choline esterase genes amplification in organophosphate resistance evolution in Ae. aegypti, reveals the diversity among individuals and populations of this resistance mechanism, because of variation both in the identity of the genes amplified and in their copy number and sets up a fast and efficient tool to detect and follow the spread of these resistant alleles in the field. Additionally, the different experimental approaches adopted have generated genomic and transcriptomic data, of which only the part related to CCE gene amplification has been exploited. These data are very likely to reveal other genomic and expression determinants of resistance that will give access to an extra degree of complexity in organophosphate insecticide resistance determinism and evolution.
References
Bass C, Field LM (2011) Gene amplification and insecticide resistance. Pest Management Science, 67, 886–890. https://doi.org/10.1002/ps.2189
Cattel J, Haberkorn C, Laporte F, Gaude T, Cumer T, Renaud J, Sutherland IW, Hertz JC, Bonneville J-M, Arnaud V, Nous C, Fustec B, Boyer S, Marcombe S, David J-P (2020) A genomic amplification affecting a carboxylesterase gene cluster confers organophosphate resistance in the mosquito Aedes aegypti: from genomic characterization to high-throughput field detection. bioRxiv, 2020.06.08.139741, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. https://doi.org/10.1101/2020.06.08.139741
Moyes CL, Vontas J, Martins AJ, Ng LC, Koou SY, Dusfour I, Raghavendra K, Pinto J, Corbel V, David J-P, Weetman D (2017) Contemporary status of insecticide resistance in the major Aedes vectors of arboviruses infecting humans. PLOS Neglected Tropical Diseases, 11, e0005625. https://doi.org/10.1371/journal.pntd.0005625
| A genomic amplification affecting a carboxylesterase gene cluster confers organophosphate resistance in the mosquito Aedes aegypti: from genomic characterization to high-throughput field detection | Julien Cattel, Chloé Haberkorn, Fréderic Laporte, Thierry Gaude, Tristan Cumer, Julien Renaud, Ian W. Sutherland, Jeffrey C. Hertz, Jean-Marc Bonneville, Victor Arnaud, Camille Noûs, Bénédicte Fustec, Sébastien Boyer, Sébastien Marcombe, Jean-Phil... | <p>By altering gene expression and creating paralogs, genomic amplifications represent a key component of short-term adaptive processes. In insects, the use of insecticides can select gene amplifications causing an increased expression of detoxifi... | 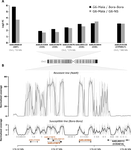 | Adaptation, Evolutionary Applications, Experimental Evolution, Genome Evolution, Molecular Evolution | Stephanie Bedhomme | | 2020-06-09 13:27:18 | View |





