An evolutionary view of a biomedically important gene family
Recommended by Kateryna Makova based on reviews by 2 anonymous reviewers
This manuscript [1] investigates the evolutionary history of the DAN gene family—a group of genes important for embryonic development of limbs, kidneys, and left-right axis speciation. This gene family has also been implicated in a number of diseases, including cancer and nephropathies. DAN genes have been associated with the inhibition of the bone morphogenetic protein (BMP) signaling pathway. Despite this detailed biochemical and functional knowledge and clear importance for development and disease, evolution of this gene family has remained understudied. The diversification of this gene family was investigated in all major groups of vertebrates. The monophyly of the gene members belonging to this gene family was confirmed. A total of five clades were delineated, and two novel lineages were discovered. The first lineage was only retained in cephalochordates (amphioxus), whereas the second one (GREM3) was retained by cartilaginous fish, holostean fish, and coelanth. Moreover, the patterns of chromosomal synteny in the chromosomal regions harboring DAN genes were investigated. Additionally, the authors reconstructed the ancestral gene repertoires and studied the differential retention/loss of individual gene members across the phylogeny. They concluded that the ancestor of gnathostome vertebrates possessed eight DAN genes that underwent differential retention during the evolutionary history of this group. During radiation of vertebrates, GREM1, GREM2, SOST, SOSTDC1, and NBL1 were retained in all major vertebrate groups. At the same time, GREM3, CER1, and DAND5 were differentially lost in some vertebrate lineages. At least two DAN genes were present in the common ancestor of vertebrates, and at least three DAN genes were present in the common ancestor of chordates. Therefore the patterns of retention and diversification in this gene family appear to be complex. Evolutionary slowdown for the DAN gene family was observed in mammals, suggesting selective constraints. Overall, this article puts the biomedical importance of the DAN family in the evolutionary perspective.
References
[1] Opazo JC, Hoffmann FG, Zavala K, Edwards SV (2020) Evolution of the DAN gene family in vertebrates. bioRxiv, 794404, ver. 3 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/794404
| Evolution of the DAN gene family in vertebrates | Juan C. Opazo, Federico G. Hoffmann, Kattina Zavala, Scott V. Edwards | <p>The DAN gene family (DAN, Differential screening-selected gene Aberrant in Neuroblastoma) is a group of genes that is expressed during development and plays fundamental roles in limb bud formation and digitation, kidney formation and morphogene... | 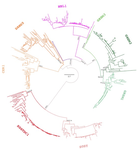 | Molecular Evolution | Kateryna Makova | | 2019-10-15 16:43:13 | View |
Transcriptional differences between the two host strains of Spodoptera frugiperda (Lepidoptera: Noctuidae)
Marion Orsucci, Yves Moné, Philippe Audiot, Sylvie Gimenez, Sandra Nhim, Rima Naït-Saïdi, Marie Frayssinet, Guillaume Dumont, Jean-Paul Boudon, Marin Vabre, Stéphanie Rialle, Rachid Koual, Gael J. Kergoat, Rodney N. Nagoshi, Robert L. Meagher, Emmanuelle d'Alencon, Nicolas Nègre
https://doi.org/10.1101/263186
Speciation through selection on mitochondrial genes?
Recommended by Astrid Groot based on reviews by Heiko Vogel and Sabine Haenniger
Whether speciation through ecological specialization occurs has been a thriving research area ever since Mayr (1942) stated this to play a central role. In herbivorous insects, ecological specialization is most likely to happen through host plant differentiation (Funk et al. 2002). Therefore, after Dorothy Pashley had identified two host strains in the Fall armyworm (FAW), Spodoptera frugiperda, in 1988 (Pashley 1988), researchers have been trying to decipher the evolutionary history of these strains, as this seems to be a model species in which speciation is currently occurring through host plant specialization. Even though FAW is a generalist, feeding on many different host plant species (Pogue 2002) and a devastating pest in many crops, Pashley identified a so-called corn strain and a so-called rice strain in Puerto Rico. Genetically, these strains were found to differ mostly in an esterase, although later studies showed additional genetic differences and markers, mostly in the mitochondrial COI and the nuclear TPI. Recent genomic studies showed that the two strains are overall so genetically different (2% of their genome being different) that these two strains could better be called different species (Kergoat et al. 2012). So far, the most consistent differences between the strains have been their timing of mating activities at night (Schoefl et al. 2009, 2011; Haenniger et al. 2019) and hybrid incompatibilities (Dumas et al. 2015; Kost et al. 2016). Whether and to what extent host plant preference or performance contributed to the differentiation of these sympatrically occurring strains has remained unclear.
In the current study, Orsucci et al. (2020) performed oviposition assays and reciprocal transplant experiments with both strains to measure fitness effects, in combination with a comprehensive RNAseq experiment, in which not only lab reared larvae were analysed, but also field-collected larvae. When testing preference and performance on the two host plants corn and rice, the authors did not find consistent fitness differences between the two strains, with both strains performing less on rice plants, although larvae from the corn strain survived more on corn plants than those from the rice strain. These results mostly confirm findings of a number of investigations over the past 30 years, where no consistent differences on the two host plants were observed (reviewed in Groot et al. 2016). However, the RNAseq experiments did show some striking differences between the two strains, especially in the reciprocally transplanted larvae, where both strains had been reared on rice or on corn plants for one generation: both strains showed transcriptional responses that correspond to their respective putative host plants, i.e. overexpression of genes involved in digestion and metabolic activity, and underexpression of genes involved in detoxification, in the corn strain on corn and in the rice strain on rice. Interestingly, similar sets of genes were found to be overexpressed in the field-collected larvae with which a RNAseq experiment was conducted as well.
The most interesting result of the study performed by Orsucci et al. (2020) is the underexpression in the corn strain of so-called numts, small genomic sequences that corresponded to fragments of the mitochondrial COI and COIII. These two numts were differentially expressed in the two strains in all RNAseq experiments analysed. This result coincides with the fact that the COI is one of the main diagnostic markers to distinguish these two strains. The authors suggestion that a difference in energy production between these two strains may be linked to a shift in host plant preference matches their finding that rice plants seem to be less suitable host plants than corn plants. However, as the lower suitability of rice plants was true for both strains, it remains unclear whether and how this difference could be linked to possible host plant differentiation between the strains. The authors also suggest that COI and potentially other mitochondrial genes may be the original target of selection between these two strains. This is especially interesting in light of the fact that field-collected larvae have frequently been found to have a rice strain mitochondrial genotype and a corn strain nuclear genotype, also in this study, while in the lab (female rice strain x male corn strain) hybrid females (i.e. females with a rice strain mitochondrial genotype and a corn strain nuclear genotype) are behaviorally sterile (Kost et al. 2016). Whether and how selection on mitochondrial genes or on mitonuclear interactions has indeed affected the evolution of these strains in the New world, and will affect the evolution of FAW in newly invaded habitats in the Old world, including Asia and Australia – where, so far, only corn strain and (female rice strain x male corn strain) hybrids have been found (Nagoshi 2019), will be a challenging research question for the coming years.
References
[1] Dumas, P. et al. (2015). Spodoptera frugiperda (Lepidoptera: Noctuidae) host-plant variants: two host strains or two distinct species?. Genetica, 143(3), 305-316. doi: 10.1007/s10709-015-9829-2
[2] Funk, D. J., Filchak, K. E. and Feder J. L. (2002) Herbivorous insects: model systems for the comparative study of speciation ecology. In: Etges W.J., Noor M.A.F. (eds) Genetics of Mate Choice: From Sexual Selection to Sexual Isolation. Contemporary Issues in Genetics and Evolution, vol 9. Springer, Dordrecht. doi: 10.1007/978-94-010-0265-3_10
[3] Groot, A. T., Unbehend, M., Hänniger, S., Juárez, M. L., Kost, S. and Heckel D. G.(2016) Evolution of reproductive isolation of Spodoptera frugiperda. In Allison, J. and Cardé, R. (eds) Sexual communication in moths. Chapter 20: 291-300.
[4] Hänniger, S. et al. (2017). Genetic basis of allochronic differentiation in the fall armyworm. BMC evolutionary biology, 17(1), 68. doi: 10.1186/s12862-017-0911-5
[5] Kost, S., Heckel, D. G., Yoshido, A., Marec, F., and Groot, A. T. (2016). AZ‐linked sterility locus causes sexual abstinence in hybrid females and facilitates speciation in Spodoptera frugiperda. Evolution, 70(6), 1418-1427. doi: 10.1111/evo.12940
[6] Mayr, E. (1942) Systematics and the origin of species. Columbia University Press, New York.
[7] Nagoshi, R. N. (2019). Evidence that a major subpopulation of fall armyworm found in the Western Hemisphere is rare or absent in Africa, which may limit the range of crops at risk of infestation. PloS one, 14(4). doi: 10.1371/journal.pone.0208966
[8] Orsucci, M., Moné, Y., Audiot, P., Gimenez, S., Nhim, S., Naït-Saïdi, R., Frayssinet, M., Dumont, G., Boudon, J.-P., Vabre, M., Rialle, S., Koual, R., Kergoat, G. J., Nagoshi, R. N., Meagher, R. L., d’Alençon, E. and Nègre N. (2020) Transcriptional differences between the two host strains of Spodoptera frugiperda (Lepidoptera: Noctuidae). bioRxiv, 263186, ver. 2 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/263186
[9] Pashley, D. P. (1988) Current Status of Fall Armyworm Host Strains. Florida Entomologist 71 (3): 227–34. doi: 10.2307/3495425
[10] Pogue, M. (2002). A World Revision of the Genus Spodoptera Guenée (Lepidoptera: Noctuidae). American Entomological Society.
[11] Schöfl, G., Heckel, D. G., and Groot, A. T. (2009). Time‐shifted reproductive behaviours among fall armyworm (Noctuidae: Spodoptera frugiperda) host strains: evidence for differing modes of inheritance. Journal of Evolutionary Biology, 22(7), 1447-1459. doi: 10.1111/j.1420-9101.2009.01759.x
[12] Schöfl, G., Dill, A., Heckel, D. G., and Groot, A. T. (2011). Allochronic separation versus mate choice: nonrandom patterns of mating between fall armyworm host strains. The American Naturalist, 177(4), 470-485. doi: 10.1086/658904
| Transcriptional differences between the two host strains of Spodoptera frugiperda (Lepidoptera: Noctuidae) | Marion Orsucci, Yves Moné, Philippe Audiot, Sylvie Gimenez, Sandra Nhim, Rima Naït-Saïdi, Marie Frayssinet, Guillaume Dumont, Jean-Paul Boudon, Marin Vabre, Stéphanie Rialle, Rachid Koual, Gael J. Kergoat, Rodney N. Nagoshi, Robert L. Meagher, Emm... | <p>Spodoptera frugiperda, the fall armyworm (FAW), is an important agricultural pest in the Americas and an emerging pest in sub-Saharan Africa, India, East-Asia and Australia, causing damage to major crops such as corn, sorghum and soybean. While... |  | Adaptation, Evolutionary Ecology, Expression Studies, Life History, Speciation | Astrid Groot | | 2018-05-09 13:04:34 | View |
Molecular evolution through the joint lens of genomic and population processes.
Recommended by Guillaume Achaz based on reviews by Benoit Nabholz and 1 anonymous reviewer
In their perspective article, F Pouyet and KJ Gilbert (2020), propose an interesting overview of all the processes that sculpt patterns of molecular evolution. This well documented article covers most (if not all) important facets of the recurrent debate that has marked the history of molecular evolution: the relative importance of natural selection and neutral processes (i.e. genetic drift). I particularly enjoyed reading this review, that instead of taking a clear position on the debate, catalogs patiently every pieces of information that can help understand how patterns we observed at the genome level, can be understood from a selectionnist point of view, from a neutralist one, and, to quote their title, from "everything in between". The review covers the classical objects of interest in population genetics (genetic drift, selection, demography and structure) but also describes several genomic processes (meiotic drive, linked selection, gene conversion and mutation processes) that obscure the interpretation of these population processes. The interplay between all these processes is very complex (to say the least) and have resulted in many cases in profound confusions while analyzing data. It is always very hard to fully acknowledge our ignorance and we have many times payed the price of model misspecifications. This review has the grand merit to improve our awareness in many directions. Being able to cover so many aspects of a wide topic, while expressing them simply and clearly, connecting concepts and observations from distant fields, is an amazing "tour de force". I believe this article constitutes an excellent up-to-date introduction to the questions and problems at stake in the field of molecular evolution and will certainly also help established researchers by providing them a stimulating overview supported with many relevant references.
References
[1] Pouyet F, Gilbert KJ (2020) Towards an improved understanding of molecular evolution: the relative roles of selection, drift, and everything in between. arXiv:1909.11490 [q-bio]. ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. url:https://arxiv.org/abs/1909.11490
| Towards an improved understanding of molecular evolution: the relative roles of selection, drift, and everything in between | Fanny Pouyet and Kimberly J. Gilbert | <p>A major goal of molecular evolutionary biology is to identify loci or regions of the genome under selection versus those evolving in a neutral manner. Correct identification allows accurate inference of the evolutionary process and thus compreh... | 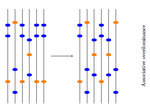 | Genome Evolution, Population Genetics / Genomics | Guillaume Achaz | | 2019-09-26 10:58:10 | View |
Further questions on the meaning of effective population size
Recommended by Martin Lascoux based on reviews by 3 anonymous reviewers
In spite of its name, the effective population size, Ne, has a complex and often distant relationship to census population size, as we usually understand it. In truth, it is primarily an abstract concept aimed at measuring the amount of genetic drift occurring in a population at any given time. The standard way to model random genetic drift in population genetics is the Wright-Fisher model and, with a few exceptions, definitions of the effective population size stems from it: “a certain model has effective population size, Ne, if some characteristic of the model has the same value as the corresponding characteristic for the simple Wright-Fisher model whose actual size is Ne” (Ewens 2004). Since Sewall Wright introduced the concept of effective population size in 1931 (Wright 1931), it has flourished and there are today numerous definitions of it depending on the process being examined (genetic diversity, loss of alleles, efficacy of selection) and the characteristic of the model that is considered. These different definitions of the effective population size were generally introduced to address specific aspects of the evolutionary process. One aspect that has been hotly debated since the first estimates of genetic diversity in natural populations were published is the so-called Lewontin’s paradox (1974). Lewontin noted that the observed variation in heterozygosity across species was much smaller than one would expect from the neutral expectations calculated with the actual size of the species.
In essence, what Galtier and Rousselle propose in their clever paper is to introduce a new approach to compare effective population sizes across species and thereby a new way to address Lewontin’s paradox. Classically, the effective population size in this type of comparative genomic studies is simply estimated from nucleotide diversity at putatively neutral sites using the equation relating levels of diversity (θ) to mutation rate per generation (μ) and effective population size, Ne, θ = 4Neμ. As Galtier and Rousselle point out there are many issues with this approach. In particular, although we can now estimate θ very precisely, we generally do not have a reliable estimate of the mutation rate, and the method rests on many, unwarranted, assumptions; for example that the population is at mutation-drift equilibrium. Instead they propose to estimate the effective population size from the load of segregating deleterious mutations which can be summarized by the ratio of nonsynonymous to synonymous mutations, πN/πS: small-Ne species are expected to accumulate more deleterious mutations and carry a higher load than large-Ne ones at selection/drift equilibrium (Ohta et al. 1973; Welch et al. 2008). At first glance, this suggestion seems counterintuitive since considering sites under selection undoubtedly adds a new layer of complexity to an already intricate situation. Indeed, one is now bringing to the brew another elusive object, namely, the Distribution of Fitness Effect of mutations (DFE). However, estimating Ne from the load of segregating deleterious mutations may actually simplify the situation in two important ways. First, using πN/πS does not require assumption about μ (as the mutation rate will cancel out in the ratio). Second, the ratio of nonsynonymous to synonymous nucleotide diversity, reaches equilibrium faster than the nucleotide diversity at synonymous site after a change in population size, so we can hope for less sensitivity to (often unknown) recent demographic history (Brandvain and Wright 2016).
Extending recent developments in the estimation of the DFE, Galtier and Rousselle eventually obtain estimates of the average deleterious effect, 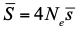 , where Ne is the effective population size and , where Ne is the effective population size and  is the mean fitness effect of non-synonymous mutations. Assuming further that distinct species share a common DFE and therefore a common , they obtain estimates of the between species ratio of Ne from the between species ratio of is the mean fitness effect of non-synonymous mutations. Assuming further that distinct species share a common DFE and therefore a common , they obtain estimates of the between species ratio of Ne from the between species ratio of  . Applying their newly developed approach to various datasets they conclude that the power of drift varies by a factor of at least 500 between large-Ne (Drosophila) and small-Ne species (H. sapiens). This is an order of magnitude larger than what would be obtained by comparing estimates of the variation in neutral diversity. Hence the proposed approach seems to have gone some way in making Lewontin’s paradox less paradoxical. But, perhaps more importantly, as the authors tersely point out at the end of the abstract their results further questions the meaning of Ne parameters in population genetics. And arguably this could well be the most important contribution of their study and something that is badly needed. . Applying their newly developed approach to various datasets they conclude that the power of drift varies by a factor of at least 500 between large-Ne (Drosophila) and small-Ne species (H. sapiens). This is an order of magnitude larger than what would be obtained by comparing estimates of the variation in neutral diversity. Hence the proposed approach seems to have gone some way in making Lewontin’s paradox less paradoxical. But, perhaps more importantly, as the authors tersely point out at the end of the abstract their results further questions the meaning of Ne parameters in population genetics. And arguably this could well be the most important contribution of their study and something that is badly needed.
References
Brandvain Y, Wright SI (2016) The Limits of Natural Selection in a Nonequilibrium World. Trends in Genetics, 32, 201–210. doi: 10.1016/j.tig.2016.01.004
Ewens WJ (2010) Mathematical Population Genetics: Theoretical Introduction. Springer-Verlag New York Inc., New York, NY. doi: 10.1007/978-0-387-21822-9
Galtier N, Rousselle M (2020) How much does Ne vary among species? bioRxiv, 861849, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/861849
Lewontin RC (1974) The genetic basis of evolutionary change. Columbia University Press, New York.
Ohta T (1973) Slightly Deleterious Mutant Substitutions in Evolution. Nature, 246, 96–98. doi: 10.1038/246096a0
Welch JJ, Eyre-Walker A, Waxman D (2008) Divergence and Polymorphism Under the Nearly Neutral Theory of Molecular Evolution. Journal of Molecular Evolution, 67, 418–426. doi: 10.1007/s00239-008-9146-9
Wright S (1931) Evolution in Mendelian Populations. Genetics, 16, 97–159. url: https://www.genetics.org/content/16/2/97
| How much does Ne vary among species? | Nicolas Galtier, Marjolaine Rousselle | <p>Genetic drift is an important evolutionary force of strength inversely proportional to *Ne*, the effective population size. The impact of drift on genome diversity and evolution is known to vary among species, but quantifying this effect is a d... | 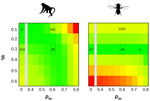 | Bioinformatics & Computational Biology, Genome Evolution, Molecular Evolution, Population Genetics / Genomics | Martin Lascoux | | 2019-12-08 00:11:00 | View |
The insertion of a mitochondrial selfish element into the nuclear genome and its consequences
Julien Y. Dutheil, Karin Münch, Klaas Schotanus, Eva H. Stukenbrock and Regine Kahmann
https://doi.org/10.1101/787044
Some evolutionary insights into an accidental homing endonuclease passage from mitochondria to the nucleus
Recommended by Sylvain Charlat based on reviews by Jan Engelstaedter and Yannick Wurm
Not all genetic elements composing genomes are there for the benefit of their carrier. Many have no consequences on fitness, or too mild ones to be eliminated by selection, and thus stem from neutral processes. Many others are indeed the product of selection, but one acting at a different level, increasing the fitness of some elements of the genome only, at the expense of the “organism” as a whole. These can be called selfish genetic elements, and come into a wide variety of flavours [1], illustrating many possible means to cheat with “fair” reproductive processes such as meiosis, and thus get overrepresented in the offspring of their hosts. Producing copies of itself through transposition is one such strategy; a very successful one indeed, explaining a large part of the genomic content of many organisms. Killing non carrier gametes following meiosis in heterozygous carriers is another one. Less know and less common is the ability of some elements to turn heterozygous carriers into homozygous ones, that will thus transmit the selfish elements to all offspring instead of half. This is achieved by nucleic sequences encoding so-called “Homing endonucleases” (HEs). These proteins tend to induce double strand breaks of DNA specifically in regions homologous to their own insertion sites. The recombination machinery is such that the intact homologous region, that is, the one carrying the HE sequence, is then used as a template for the reparation of the break, resulting in the effective conversion of a non-carrier allele into a carrier allele. Such elements can also occur in the mitochondrial genomes of organisms where mitochondria are not strictly transmitted by one parent only, offering mitochondrial HEs some opportunities for “homing” into new non carrier genomes. This is the case in yeasts, where HEs were first reported [2,3].
In this new study, based on genomic experimental data from the fungal maize pathogen Ustilago maydis, Julien Dutheil and colleagues [4] document one possible evolutionary pathway for which little evidence existed before: the passage of a mitochondrial HE into the nuclear genome. The GC content of this region leaves little doubt on its mitochondrial origin, and homologs can indeed be found in the mitochondrial genomes of close relatives. Strangely enough, U. maydis itself does not appear to carry this selfish element in its own mitochondria, suggesting it may have been acquired from a different species, or be subject to a sufficiently rapid turnover to have been recently lost.
Many elements of the story uncovered by this study remain mysterious. How, in the first place, was this HE gene inserted in a nuclear genomic region that shows no apparent homology with its original insertion site, making typical “homing” a not-so-likely explanation? This question may in fact be generalised to many HE systems: is the first insertion into a homing site always the product of a typical homing event, which implies the presence of an homologous template DNA fragment, or can HE genes insert through other means? But then, why specifically in regions that would be targeted by the nuclease they encode? What is the evolutionary fate of this newly inserted element? The new gene may well be on its way to pseudogenisation, as suggested by the truncation of its upper part, precluding its functioning as a HE, and the lack of evidence of selective constraints through dN/dS analysis; but the mutation generated by the insertion event may have phenotypic implications, possibly through the partial truncation of another gene, encoding a helicase. How old is this insertion? The fact that it has accumulated some mutations makes a very recent event rather unlikely, but this insertion has been detected in only one isolate of U. maydis, suggesting it is not so frequent in natural populations.
Whatever the answers to these open questions, that will hopefully be addressed by further work on this system, the present study has revealed that horizontal transmission enlarges the scope of possible evolutionary consequences of HE genes, that may move not only between mitochondrial genomes, but also occasionally into a nucleus.
References
[1] Burt, A., and Trivers, R. (2006). Genes in Conflict: The Biology of Selfish Genetic Elements. Belknap Press.
[2] Coen, D., Deutch, J., Netter, P., Petrochillo, E., and Slonimski, P. (1970). Mitochondrial genetics. I. Methodology and phenomenology. Symposia of the Society for Experimental Biology, 24, 449-496.
[3] Colleaux, L., D’Auriol, L., Betermier, M., Cottarel, G., Jacquier, A., Galibert, F., and Dujon, B. (1986). Universal code equivalent of a yeast mitochondrial intron reading frame is expressed into E. coli as a specific double strand endonuclease. Cell, 44, 521–533. doi: 10.1016/0092-8674(86)90262-X
[4] Dutheil, J. Y., Münch, K., Schotanus, K., Stukenbrock, E. H., and Kahmann, R. (2020). The insertion of a mitochondrial selfish element into the nuclear genome and its consequences. bioRxiv, 787044, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/787044
| The insertion of a mitochondrial selfish element into the nuclear genome and its consequences | Julien Y. Dutheil, Karin Münch, Klaas Schotanus, Eva H. Stukenbrock and Regine Kahmann | <p>Homing endonucleases (HE) are enzymes capable of cutting DNA at highly specific target sequences, the repair of the generated double-strand break resulting in the insertion of the HE-encoding gene ("homing" mechanism). HEs are present in all th... |  | Genome Evolution, Molecular Evolution | Sylvain Charlat | | 2019-09-30 20:34:23 | View |
Potential adaptive divergence between subspecies and populations of snapdragon plants inferred from QST – FST comparisons
Sara Marin, Anaïs Gibert, Juliette Archambeau, Vincent Bonhomme, Mylène Lascoste and Benoit Pujol
https://doi.org/10.5281/zenodo.3628168
From populations to subspecies… to species? Contrasting patterns of local adaptation in closely-related taxa and their potential contribution to species divergence
Recommended by Emmanuelle Porcher based on reviews by Sophie Karrenberg, Santiago C. Gonzalez-Martinez and 1 anonymous reviewer
Elevation gradients are convenient and widely used natural setups to study local adaptation, particularly in these times of rapid climate change [e.g. 1]. Marin and her collaborators [2] did not follow the mainstream, however. Instead of tackling adaptation to climate change, they used elevation gradients to address another crucial evolutionary question [3]: could adaptation to altitude lead to ecological speciation, i.e. reproductive isolation between populations in spite of gene flow? More specifically, they examined how much local adaptation to environmental variation differed among closely-related, recently diverged subspecies. They studied several populations of two subspecies of snapdragon (Antirrhinum majus), with adjacent geographical distributions. Using common garden experiments and the classical, but still useful, QST-FST comparison, they demonstrate contrasting patterns of local adaptation to altitude between the two subspecies, with several traits under divergent selection in A. majus striatum but none in A. majus pseudomajus. These differences in local adaptation may contribute to species divergence, and open many stimulating questions on the underlying mechanisms, such as the identity of environmental drivers or contribution of reproductive isolation involving flower color polymorphism.
References
[1] Anderson, J. T., and Wadgymar, S. M. (2020). Climate change disrupts local adaptation and favours upslope migration. Ecology letters, 23(1), 181-192. doi: 10.1111/ele.13427
[2] Marin, S., Gibert, A., Archambeau, J., Bonhomme, V., Lascoste, M., and Pujol, B. (2020). Potential adaptive divergence between subspecies and populations of snapdragon plants inferred from QST – FST comparisons. Zenodo, 3628168, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. doi: 10.5281/zenodo.3628168
[3] Schluter, D. (2009). Evidence for ecological speciation and its alternative. Science, 323(5915), 737-741. doi: 10.1126/science.1160006
| Potential adaptive divergence between subspecies and populations of snapdragon plants inferred from QST – FST comparisons | Sara Marin, Anaïs Gibert, Juliette Archambeau, Vincent Bonhomme, Mylène Lascoste and Benoit Pujol | <p>Phenotypic divergence among natural populations can be explained by natural selection or by neutral processes such as drift. Many examples in the literature compare putatively neutral (FST) and quantitative genetic (QST) differentiation in mult... |  | Adaptation, Evolutionary Ecology, Genotype-Phenotype, Morphological Evolution, Quantitative Genetics | Emmanuelle Porcher | | 2018-08-05 15:34:30 | View |
Meta-population structure and the evolutionary transition to multicellularity
Caroline J Rose, Katrin Hammerschmidt, Yuriy Pichugin and Paul B Rainey
https://doi.org/10.1101/407163
The ecology of evolutionary transitions to multicellularity
Recommended by Dustin Brisson based on reviews by 2 anonymous reviewers
The evolutionary transition to multicellular life from free-living, single-celled ancestors has occurred independently in multiple lineages [1-5]. This evolutionary transition to cooperative group living can be difficult to explain given the fitness advantages enjoyed by the non-cooperative, single-celled organisms that still numerically dominate life on earth [1,6,7]. Although several hypotheses have been proposed to explain the transition to multicellularity, a common theme is the abatement of the efficacy of natural selection among the single cells during the free-living stage and the promotion of the efficacy of selection among groups of cells during the cooperative stage, an argument reminiscent of those from George Williams’ seminal book [8,9]. The evolution of life cycles appears to be a key step in the transition to multicellularity as it can align fitness advantages of the single-celled 'reproductive' stage with that of the cooperative 'organismal' stage [9-12]. That is, the evolution of life cycles allows natural selection to operate over timescales longer than that of the doubling time of the free-living cells [13]. Despite the importance of this issue, identifying the range of ecological conditions that reduce the importance of natural selection at the single-celled, free-living stage and increase the importance of selection among groups of cooperating cells has not been addressed empirically.
Rose et al [14] addressed this issue in a series of real time evolution experiments with bacteria in which they varied the intensity of between-group versus individual-level selection. Central to the experiment is an ecological scaffold that requires lineages to switch between free-living (reproductive) and group-living (organismal) life-stages. One ecological scenario severely limited natural selection at the single-celled, free-living stage by maintaining separation among the reproductive propagules originating from different organisms (groups of cells derived from a single ancestral cell). A second ecological scenario mixed the reproductive propagules from different organisms, leading to severe competition between single cells derived from both the same and other 'organisms'. These ecological scenarios lead to very different evolutionary outcomes. Limiting competition, and thus natural selection, at the reproductive propagule stage promoted traits that favored organismal fitness at the expense of cell division, while competition among single-cells favored traits that promote cell-level traits at the expense of group-level traits. The authors investigate a range of measures of cell and group-level performance in order to understand the mechanisms favoring organismal versus single-cell fitness. Importantly, an evolutionary trade-off between traits promoting organismal fitness and single-cell fitness appears to constrain maximizing fitness of both phases, especially when strong natural selection acts on the single-cell stage.
This article is incredibly thorough and utilizes multiple experiments and levels of argument in order to support the conclusions. The authors include considerable discussion of broader topics surrounding the immediate hypotheses throughout the article, which add both clarity and complexity. The complexity of the experiments, results, and the topic itself lead to a thought-heavy article in a throwback to the monographs of old; expect to read each section multiple times.
References
[1] Maynard Smith, J. and Szathmáry, E. (1995). The Major Transitions in Evolution. Oxford, UK: Freeman. p 346.
[2] Bonner, J. T. (1998). The origins of multicellularity. Integrative Biology: Issues, News, and Reviews: Published in Association with The Society for Integrative and Comparative Biology, 1(1), 27-36. doi: 10.1002/(SICI)1520-6602(1998)1:1<27::AID-INBI4>3.0.CO;2-6
[3] Kaiser, D. (2001). Building a multicellular organism. Annual review of genetics, 35(1), 103-123. doi: 10.1146/annurev.genet.35.102401.090145
[4] Medina, M., Collins, A. G., Taylor, J. W., Valentine, J. W., Lipps, J. H., Amaral-Zettler, L., and Sogin, M. L. (2003). Phylogeny of Opisthokonta and the evolution of multicellularity and complexity in Fungi and Metazoa. International Journal of Astrobiology, 2(3), 203-211. doi: 10.1017/S1473550403001551
[5] King, N. (2004). The unicellular ancestry of animal development. Developmental cell, 7(3), 313-325. doi: 10.1016/j.devcel.2004.08.010
[6] Michod R. E. (1999). Darwinian Dynamics. Evolutionary Transitions in Fitness and Individuality. Princeton, NJ: Princeton Univ. Press. p 262.
[7] Lynch, M. (2007). The frailty of adaptive hypotheses for the origins of organismal complexity. Proceedings of the National Academy of Sciences, 104(suppl 1), 8597-8604. doi: 10.1073/pnas.0702207104
[8] Williams, G. C. (1996). Adaptation and Natural Selection, Reprint edition. Princeton, NJ: Princeton Univ. Press.
[9] Grosberg, R. K., and Strathmann, R. R. (2007). The evolution of multicellularity: a minor major transition?. Annu. Rev. Ecol. Evol. Syst., 38, 621-654. doi: 10.1146/annurev.ecolsys.36.102403.114735
[10] Buss, L. W. (1987). The Evolution of Individuality. Princeton, NJ: Princeton Univ. Press.
[11] Godfrey-Smith, P. (2009). Darwinian Populations and Natural Selection. Oxford University Press, USA.
[12] Van Gestel, J., and Tarnita, C. E. (2017). On the origin of biological construction, with a focus on multicellularity. Proceedings of the National Academy of Sciences, 114(42), 11018-11026. doi: 10.1073/pnas.1704631114
[13] Black, A. J., Bourrat, P., and Rainey, P. B. (2020). Ecological scaffolding and the evolution of individuality. Nature Ecology & Evolution, 4(3), 426-436. doi: 10.1038/s41559-019-1086-9
[14] Rose, C. J., Hammerschmidt, K., Pichugin, Y. and Rainey, P. B. (2020). Meta-population structure and the evolutionary transition to multicellularity. bioRxiv, 407163, ver. 5 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/407163
| Meta-population structure and the evolutionary transition to multicellularity | Caroline J Rose, Katrin Hammerschmidt, Yuriy Pichugin and Paul B Rainey | <p>The evolutionary transition to multicellularity has occurred on numerous occasions, but transitions to complex life forms are rare. While the reasons are unclear, relevant factors include the intensity of within- versus between-group selection ... | 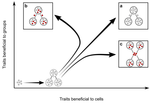 | Adaptation, Evolutionary Dynamics, Experimental Evolution | Dustin Brisson | | 2019-04-04 12:26:36 | View |
Reconciling the upsides and downsides of migration for evolutionary rescue
Recommended by Claudia Bank based on reviews by 3 anonymous reviewers based on reviews by 3 anonymous reviewers
The evolutionary response of populations to changing or novel environments is a topic that unites the interests of evolutionary biologists, ecologists, and biomedical researchers [1]. A prominent phenomenon in this research area is evolutionary rescue, whereby a population that is otherwise doomed to extinction survives due to the spread of new or pre-existing mutations that are beneficial in the new environment. Scenarios of evolutionary rescue require a specific set of parameters: the absolute growth rate has to be negative before the rescue mechanism spreads, upon which the growth rate becomes positive. However, potential examples of its relevance exist (e.g., [2]). From a theoretical point of view, the technical challenge but also the beauty of evolutionary rescue models is that they combine the study of population dynamics (i.e., changes in the size of populations) and population genetics (i.e., changes in the frequencies in the population). Together, the potential relevance of evolutionary rescue in nature and the models' theoretical appeal has resulted in a suite of modeling studies on the subject in recent years.
In this manuscript [3], Tomasini and Peischl address a question that has been contentiously discussed in the literature: when does migration favor evolutionary rescue? They expand on past work (specifically, [4, 5]) by studying the influence of the interaction of the speed and severity of environmental change and the amount of dispersal on the probability of evolutionary rescue. They develop simple analytical results (complemented by simulations) for a haploid one-locus model of two populations connected by gene flow, where both populations deteriorate successively such that evolutionary rescue is required for the metapopulation to survive. For example, the authors derive a simple analytical condition demonstrating that migration between the subpopulations favors evolutionary rescue if environmental change occurs slowly across the two populations (which leaves time for the second population to serve as an immigration source), if the new environment is very harsh and/or if rescue mutations are strongly beneficial in the new environment. The latter conditions ensure that the rescue mutations can spread easily in the new environment without much competition with immigrating, maladapted, genotypes. This result is intuitive and connects between traditional single and multiple-deme models.
Altogether, Tomasini and Peischl present an extensive theoretical study and address also the effect of various tweaks to the model assumptions, such as asymmetries in gene flow and/or carrying capacities, and the effects of different density regulation and local growth rates. They successfully made an effort to explain and interpret their results for a general audience, such that also non-theoreticians should not be afraid to take a look at this manuscript.
References
[1] Bell, G. (2017). Evolutionary Rescue. Annual Review of Ecology, Evolution, and Systematics 48(1), 605-627. doi: 10.1146/annurev-ecolsys-110316-023011
[2] Oziolor, E. M., Reid, N. M., Yair, S. et al. (2019). Adaptive introgression enables evolutionary rescue from extreme environmental pollution. Science, 364(6439), 455-457. doi: 10.1126/science.aav4155
[3] Tomasini, M. and Peischl, S. (2020) When does gene flow facilitate evolutionary rescue? bioRxiv, 622142, ver. 5 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/622142
[4] Uecker, H., Otto, S. P., and Hermisson, J. (2014). Evolutionary rescue in structured populations. The American Naturalist, 183(1), E17-E35. doi: 10.1086/673914
[5] Tomasini, M., and Peischl, S. (2018). Establishment of locally adapted mutations under divergent selection. Genetics, 209(3), 885-895. doi: 10.1534/genetics.118.301104
| When does gene flow facilitate evolutionary rescue? | Matteo Tomasini, Stephan Peischl | <p>Experimental and theoretical studies have highlighted the impact of gene flow on the probability of evolutionary rescue in structured habitats. Mathematical modelling and simulations of evolutionary rescue in spatially or otherwise structured p... |  | Evolutionary Dynamics, Evolutionary Theory, Population Genetics / Genomics | Claudia Bank | | 2019-05-22 11:12:13 | View |
How do invasion syndromes evolve? An experimental evolution approach using the ladybird Harmonia axyridis
Julien Foucaud, Ruth A. Hufbauer, Virginie Ravigné, Laure Olazcuaga, Anne Loiseau, Aurelien Ausset, Su Wang, Lian-Sheng Zang, Nicolas Lemenager, Ashraf Tayeh, Arthur Weyna, Pauline Gneux, Elise Bonnet, Vincent Dreuilhe, Bastien Poutout, Arnaud Estoup, Benoit Facon
https://doi.org/10.1101/849968
Selection on a single trait does not recapitulate the evolution of life-history traits seen during an invasion
Recommended by Inês Fragata and Ben Phillips based on reviews by 2 anonymous reviewers
Biological invasions are natural experiments, and often show that evolution can affect dynamics in important ways [1-3]. While we often think of invasions as a conservation problem stemming from anthropogenic introductions [4,5], biological invasions are much more commonplace than this, including phenomena as diverse as natural range shifts, the spread of novel pathogens, and the growth of tumors. A major question across all these settings is which set of traits determine the ability of a population to invade new space [6,7]. Traits such as: increased growth or reproductive rate, dispersal ability and ability to defend from predation often show large evolutionary shifts across invasion history [1,6,8]. Are such multi-trait shifts driven by selection on multiple traits, or a correlated response by multiple traits to selection on one? Resolving this question is important for both theoretical and practical reasons [9,10]. But despite the importance of this issue, it is not easy to perform the necessary manipulative experiments [9].
Foucaud et al. [11] tackled this issue by performing experimental evolution on source populations of the invasive ladybug Harmonia axyridis. The authors tested if selection on a single trait could generate correlated responses in other life history traits. Specifically, they used experimental evolution to impose divergent selection on female mass, and reproductive timing. After ten generations, they found that selection for weight did not affect almost any other life history trait. However, nine generations of selection for faster reproduction led to correlated phenotypic changes in developmental, reproduction and survival rate of populations, although not always in the direction we might have expected. Despite this correlated response, none of their selected lines were able to fully recapitulate the trait shifts seen in natural invasions of this species. This implies that selection during natural invasions is operating on multiple traits; a finding in agreement with our growing understanding of how selection acts during introduction and invasion [12,13].
Populations undergoing a colonization process may also be subject to a multitude of different selective pressures [14,15]. The authors expanded their work in this direction by testing whether food availability alters the observed correlations between life history traits. The pervasiveness of genotype by environment interactions observed also points to a role for multiple selective pressures in shaping the suite of life-history shifts observed in wild ladybug populations. The work from Foucaud and colleagues [11] adds to a small but growing list of important studies that use experimental evolution to investigate how life-history traits evolve, and how they evolve during invasions in particular.
References
[1] Sakai, A.K., Allendorf, F.W., Holt, J.S. et al. (2001). The population biology of invasive species. Annual review of ecology and systematics, 32(1), 305-332. doi: 10.1146/annurev.ecolsys.32.081501.114037
[2] Hairston Jr, N. G., Ellner, S. P., Geber, M. A., Yoshida, T. and Fox, J. A. (2005). Rapid evolution and the convergence of ecological and evolutionary time. Ecology letters, 8(10), 1114-1127. doi: 10.1111/j.1461-0248.2005.00812.x
[3] Chuang, A. and Peterson, C. R. (2016). Expanding population edges: theories, traits, and trade‐offs. Global change biology, 22(2), 494-512. doi: 10.1111/gcb.13107
[4] Whitney, K. D. and Gabler, C. A. (2008). Rapid evolution in introduced species,‘invasive traits’ and recipient communities: challenges for predicting invasive potential. Diversity and Distributions, 14(4), 569-580. doi: 10.1111/j.1472-4642.2008.00473.x
[5] Catullo, R. A., Llewelyn, J., Phillips, B. L. and Moritz, C. C. (2019). The Potential for Rapid Evolution under Anthropogenic Climate Change. Current Biology, 29(19), R996-R1007. doi: 10.1016/j.cub.2019.08.028
[6] Suarez, A. V. and Tsutsui, N. D. (2008). The evolutionary consequences of biological invasions. Molecular Ecology, 17(1), 351-360. doi: 10.1111/j.1365-294X.2007.03456.x
[7] Deforet, M., Carmona-Fontaine, C., Korolev, K. S. and Xavier, J. B. (2019). Evolution at the edge of expanding populations. The American Naturalist, 194(3), 291-305. doi: 10.1086/704594
[8] Phillips, B. L., Brown, G. P., and Shine, R. (2010). Life‐history evolution in range‐shifting populations. Ecology, 91(6), 1617-1627. doi: 10.1890/09-0910.1
[9] Colautti, R. I. and Lau, J. A. (2015). Contemporary evolution during invasion: evidence for differentiation, natural selection, and local adaptation. Molecular ecology, 24(9), 1999-2017. doi: 10.1111/mec.13162
[10] Szűcs, M., Melbourne, B. A., Tuff, T., Weiss‐Lehman, C. and Hufbauer, R. A. (2017). Genetic and demographic founder effects have long‐term fitness consequences for colonising populations. Ecology Letters, 20(4), 436-444. doi: 10.1111/ele.12743
[11] Foucaud, J., Hufbauer, R. A., Ravigné, V., Olazcuaga, L., Loiseau, A., Ausset, A., Wang, S., Zang, L.-S., Lemenager, N., Tayeh, A., Weyna, A., Gneux, P., Bonnet, E., Dreuilhe, V., Poutout, B., Estoup, A. and Facon, B. (2020). How do invasion syndromes evolve? An experimental evolution approach using the ladybird Harmonia axyridis. bioRxiv, 849968 ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/849968
[12] Simons, A. M. (2003). Invasive aliens and sampling bias. Ecology Letters, 6(4), 278-280. doi: 10.1046/j.1461-0248.2003.00430.x
[13] Phillips, B. L. and Perkins, T. A. (2019). Spatial sorting as the spatial analogue of natural selection. Theoretical Ecology, 12(2), 155-163. doi: 10.1007/s12080-019-0412-9
[14] Lavergne, S. and Molofsky, J. (2007). Increased genetic variation and evolutionary potential drive the success of an invasive grass. Proceedings of the National Academy of Sciences, 104(10), 3883-3888. doi: 10.1073/pnas.0607324104
[15] Moran, E. V. and Alexander, J. M. (2014). Evolutionary responses to global change: lessons from invasive species. Ecology Letters, 17(5), 637-649. doi: 10.1111/ele.12262
| How do invasion syndromes evolve? An experimental evolution approach using the ladybird Harmonia axyridis | Julien Foucaud, Ruth A. Hufbauer, Virginie Ravigné, Laure Olazcuaga, Anne Loiseau, Aurelien Ausset, Su Wang, Lian-Sheng Zang, Nicolas Lemenager, Ashraf Tayeh, Arthur Weyna, Pauline Gneux, Elise Bonnet, Vincent Dreuilhe, Bastien Poutout, Arnaud Est... | <p>Experiments comparing native to introduced populations or distinct introduced populations to each other show that phenotypic evolution is common and often involves a suit of interacting phenotypic traits. We define such sets of traits that evol... |  | Adaptation, Evolutionary Applications, Experimental Evolution, Life History, Quantitative Genetics | Inês Fragata | | 2019-11-29 07:07:00 | View |
Evolution at two time-frames: ancient and common origin of two structural variants involved in local adaptation of the European plaice (Pleuronectes platessa)
Alan Le Moan, Dorte Bekkevold, Jakob Hemmer-Hansen
https://doi.org/10.1101/662577
Genomic structural variants involved in local adaptation of the European plaice
Recommended by Maren Wellenreuther based on reviews by 3 anonymous reviewers
Awareness has been growing that structural variants in the genome of species play a fundamental role in adaptive evolution and diversification [1]. Here, Le Moan and co-authors [2] report empirical genomic-wide SNP data on the European plaice (Pleuronectes platessa) across a major environmental transmission zone, ranging from the North Sea to the Baltic Sea. Regions of high linkage disequilibrium suggest the presence of two structural variants that appear to have evolved 220 kya. These two putative structural variants show weak signatures of isolation by distance when contrasted against the rest of the genome, but the frequency of the different putative structural variants appears to co-vary in some parts of the studied range with the environment, indicating the involvement of both selective and neutral processes. This study adds to the mounting body of evidence that structural genomic variants harbour significant information that allows species to respond and adapt to the local environmental context.
References
[1] Wellenreuther, M., Mérot, C., Berdan, E., & Bernatchez, L. (2019). Going beyond SNPs: the role of structural genomic variants in adaptive evolution and species diversification. Molecular ecology, 28(6), 1203-1209. doi: 10.1111/mec.15066
[2] Le Moan, A. Bekkevold, D. & Hemmer-Hansen J. (2020). Evolution at two time-frames: ancient and common origin of two structural variants involved in local adaptation of the European plaice (Pleuronectes platessa). bioRxiv, 662577, ver. 5 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/662577
| Evolution at two time-frames: ancient and common origin of two structural variants involved in local adaptation of the European plaice (Pleuronectes platessa) | Alan Le Moan, Dorte Bekkevold, Jakob Hemmer-Hansen | <p>Changing environmental conditions can lead to population diversification through differential selection on standing genetic variation. Structural variant (SV) polymorphisms provide examples of ancient alleles that in time become associated with... |  | Adaptation, Hybridization / Introgression, Population Genetics / Genomics, Speciation | Maren Wellenreuther | | 2019-07-13 12:44:01 | View |





