
Latest recommendations
| Id | Title * | Authors * | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
02 Nov 2022

Evolution of immune genes in island birds: reduction in population sizes can explain island syndromeDemographic effects may affect adaptation to islandsRecommended by Emma Berdan based on reviews by Steven Fiddaman and 3 anonymous reviewersThe unique challenges associated with living on an island often result in organisms displaying a specific suite of traits commonly referred to as “island syndrome” (Adler and Levins, 1994; Burns, 2019; Baeckens and Van Damme, 2020). Large phenotypic shifts such as changes in size (e.g. shifts to gigantism or dwarfism, Lomolino, 2005) or coloration (Doutrelant et al., 2016) abound in the literature. However, less obvious phenotypes may also play a key role in adaptation to islands. One such trait, reduced immune function, has important implications for the future of island populations in the face of anthropogenic-induced changes. Due to lower parasite pressure caused by a less diverse and less virulent parasite population, island hosts may show a decrease in immune defenses (Beadell et al., 2006; Pérez‐Rodríguez et al., 2013). However, this hypothesis has been challenged, as many studies have found ambiguous or conflicting results (Matson, 2006; Illera et al., 2015). While most previous work has examined various immunological parameters (e.g., antibody concentrations), here, Barthe et al. (2022) take the novel approach of examining molecular signatures of immune genes. Using comparative genomic data from 34 different species of birds the authors examine the ratio of synonymous substitutions (i.e., not changing an amino acid) to non-synonymous substitutions (i.e., changing an amino acid) in innate and acquired immune genes (Pn/Ps ratio). Because population sizes on islands are lower which will affect molecular evolution, they compare these results to data from 97 control genes. Assuming relaxed selection on islands predicts that the difference between the Pn/Ps ratio of immune genes and of control genes (ΔPn/Ps) is greater in island species compared to mainland ones. As with previous work the authors found that the results differ depending on the category of immune genes. Both forms of innate defense: beta-defensins and Toll-like receptors did not show higher ΔPn/Ps for island populations. As these genes still have a higher Pn/Ps than control genes, the authors argue these results are in line with these genes being under purifying selection but lacking an “island effect”. Instead, the authors argue that demographic effects (i.e., reductions in Ne) may lead to the decreased immunity documented in other studies. In contrast, there was a reduction in Pn/Ps in MHC II genes, known to be under balancing selection. This reduction was stronger in island species and thus the authors argue that this is the only class of genes where a role for relaxed selection can be invoked. Together these results demonstrate that the changes in immunity experienced by island species are complex and that different categories of immune genes can experience different selective pressures. By including control genes in their study, they particularly highlight the importance of accounting for shifts in Ne when examining patterns of island species evolution. Hopefully, this kind of framework will be applied to other taxa to determine if these results are widespread or more specific to birds. References Adler GH, Levins R (1994) The Island Syndrome in Rodent Populations. The Quarterly Review of Biology, 69, 473–490. https://doi.org/10.1086/418744 Baeckens S, Van Damme R (2020) The island syndrome. Current Biology, 30, R338–R339. https://doi.org/10.1016/j.cub.2020.03.029 Barthe M, Doutrelant C, Covas R, Melo M, Illera JC, Tilak M-K, Colombier C, Leroy T, Loiseau C, Nabholz B (2022) Evolution of immune genes in island birds: reduction in population sizes can explain island syndrome. bioRxiv, 2021.11.21.469450, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.11.21.469450 Beadell JS, Ishtiaq F, Covas R, Melo M, Warren BH, Atkinson CT, Bensch S, Graves GR, Jhala YV, Peirce MA, Rahmani AR, Fonseca DM, Fleischer RC (2006) Global phylogeographic limits of Hawaii’s avian malaria. Proceedings of the Royal Society B: Biological Sciences, 273, 2935–2944. https://doi.org/10.1098/rspb.2006.3671 Burns KC (2019) Evolution in Isolation: The Search for an Island Syndrome in Plants. Cambridge University Press, Cambridge. https://doi.org/10.1017/9781108379953 Doutrelant C, Paquet M, Renoult JP, Grégoire A, Crochet P-A, Covas R (2016) Worldwide patterns of bird colouration on islands. Ecology Letters, 19, 537–545. https://doi.org/10.1111/ele.12588 Illera JC, Fernández-Álvarez Á, Hernández-Flores CN, Foronda P (2015) Unforeseen biogeographical patterns in a multiple parasite system in Macaronesia. Journal of Biogeography, 42, 1858–1870. https://doi.org/10.1111/jbi.12548 Lomolino MV (2005) Body size evolution in insular vertebrates: generality of the island rule. Journal of Biogeography, 32, 1683–1699. https://doi.org/10.1111/j.1365-2699.2005.01314.x Matson KD (2006) Are there differences in immune function between continental and insular birds? Proceedings of the Royal Society B: Biological Sciences, 273, 2267–2274. https://doi.org/10.1098/rspb.2006.3590 Pérez-Rodríguez A, Ramírez Á, Richardson DS, Pérez-Tris J (2013) Evolution of parasite island syndromes without long-term host population isolation: parasite dynamics in Macaronesian blackcaps Sylvia atricapilla. Global Ecology and Biogeography, 22, 1272–1281. https://doi.org/10.1111/geb.12084 | Evolution of immune genes in island birds: reduction in population sizes can explain island syndrome | Mathilde BARTHE, Claire DOUTRELANT, Rita COVAS, Martim MELO, Juan Carlos ILLERA, Marie-Ka TILAK, Constance COLOMBIER, Thibault LEROY , Claire LOISEAU , Benoit NABHOLZ | <p style="text-align: justify;">Shared ecological conditions encountered by species that colonize islands often lead to the evolution of convergent phenotypes, commonly referred to as “island syndrome”. Reduced immune functions have been previousl... | | Adaptation, Molecular Evolution, Population Genetics / Genomics | Emma Berdan | 2021-11-28 11:01:31 | ||
31 Oct 2022

Genotypic sex shapes maternal care in the African Pygmy mouse, Mus minutoidesEffect of sex chromosomes on mammalian behaviour: a case study in pygmy miceRecommended by Gabriel Marais and Trine Bilde based on reviews by Marion Anne-Lise Picard, Caroline Hu and 1 anonymous reviewer based on reviews by Marion Anne-Lise Picard, Caroline Hu and 1 anonymous reviewer
In mammals, it is well documented that sexual dimorphism and in particular sex differences in behaviour are fine-tuned by gonadal hormonal profiles. For example, in lemurs, where female social dominance is common, the level of testosterone in females is unusually high compared to that of other primate females (Petty and Drea 2015). Recent studies however suggest that gonadal hormones might not be the only biological factor involved in establishing sexual dimorphism, sex chromosomes might also play a role. The four core genotype (FCG) model and other similar systems allowing to decouple phenotypic and genotypic sex in mice have provided very convincing evidence of such a role (Gatewood et al. 2006; Arnold and Chen 2009; Arnold 2020a, 2020b). This however is a new field of research and the role of sex chromosomes in establishing sexually dimorphic behaviours has not been studied very much yet. Moreover, the FCG model has some limits. Sry, the male determinant gene on the mammalian Y chromosome might be involved in some sex differences in neuroanatomy, but Sry is always associated with maleness in the FCG model, and this potential role of Sry cannot be studied using this system. Heitzmann et al. have used a natural system to approach these questions. They worked on the African Pygmy mouse, Mus minutoides, in which a modified X chromosome called X* can feminize X*Y individuals, which offers a great opportunity for elegant experiments on the effects of sex chromosomes versus hormones on behaviour. They focused on maternal care and compared pup retrieval, nest quality, and mother-pup interactions in XX, X*X and X*Y females. They found that X*Y females are significantly better at retrieving pups than other females. They are also much more aggressive towards the fathers than other females, preventing paternal care. They build nests of poorer quality but have similar interactions with pups compared to other females. Importantly, no significant differences were found between XX and X*X females for these traits, which points to an effect of the Y chromosome in explaining the differences between X*Y and other females (XX, X*X). Also, another work from the same group showed similar gonadal hormone levels in all the females (Veyrunes et al. 2022). Heitzmann et al. made a number of predictions based on what is known about the neuroanatomy of rodents which might explain such behaviours. Using cytology, they looked for differences in neuron numbers in the hypothalamus involved in the oxytocin, vasopressin and dopaminergic pathways in XX, X*X and X*Y females, but could not find any significant effects. However, this part of their work relied on very small sample sizes and they used virgin females instead of mothers for ethical reasons, which greatly limited the analysis. Interestingly, X*Y females have a higher reproductive performance than XX and X*X ones, which compensate for the cost of producing unviable YY embryos and certainly contribute to maintaining a high frequency of X* in many African pygmy mice populations (Saunders et al. 2014, 2022). X*Y females are probably solitary mothers contrary to other females, and Heitzmann et al. have uncovered a divergent female strategy in this species. Their work points out the role of sex chromosomes in establishing sex differences in behaviours. References Arnold AP (2020a) Sexual differentiation of brain and other tissues: Five questions for the next 50 years. Hormones and Behavior, 120, 104691. https://doi.org/10.1016/j.yhbeh.2020.104691 Arnold AP (2020b) Four Core Genotypes and XY* mouse models: Update on impact on SABV research. Neuroscience & Biobehavioral Reviews, 119, 1–8. https://doi.org/10.1016/j.neubiorev.2020.09.021 Arnold AP, Chen X (2009) What does the “four core genotypes” mouse model tell us about sex differences in the brain and other tissues? Frontiers in Neuroendocrinology, 30, 1–9. https://doi.org/10.1016/j.yfrne.2008.11.001 Gatewood JD, Wills A, Shetty S, Xu J, Arnold AP, Burgoyne PS, Rissman EF (2006) Sex Chromosome Complement and Gonadal Sex Influence Aggressive and Parental Behaviors in Mice. Journal of Neuroscience, 26, 2335–2342. https://doi.org/10.1523/JNEUROSCI.3743-05.2006 Heitzmann LD, Challe M, Perez J, Castell L, Galibert E, Martin A, Valjent E, Veyrunes F (2022) Genotypic sex shapes maternal care in the African Pygmy mouse, Mus minutoides. bioRxiv, 2022.04.05.487174, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.04.05.487174 Petty JMA, Drea CM (2015) Female rule in lemurs is ancestral and hormonally mediated. Scientific Reports, 5, 9631. https://doi.org/10.1038/srep09631 Saunders PA, Perez J, Rahmoun M, Ronce O, Crochet P-A, Veyrunes F (2014) Xy Females Do Better Than the Xx in the African Pygmy Mouse, Mus Minutoides. Evolution, 68, 2119–2127. https://doi.org/10.1111/evo.12387 Saunders PA, Perez J, Ronce O, Veyrunes F (2022) Multiple sex chromosome drivers in a mammal with three sex chromosomes. Current Biology, 32, 2001-2010.e3. https://doi.org/10.1016/j.cub.2022.03.029 Veyrunes F, Perez J, Heitzmann L, Saunders PA, Givalois L (2022) Separating the effects of sex hormones and sex chromosomes on behavior in the African pygmy mouse Mus minutoides, a species with XY female sex reversal. bioRxiv, 2022.07.11.499546. https://doi.org/10.1101/2022.07.11.499546 | Genotypic sex shapes maternal care in the African Pygmy mouse, Mus minutoides | Louise D Heitzmann, Marie Challe, Julie Perez, Laia Castell, Evelyne Galibert, Agnes Martin, Emmanuel Valjent, Frederic Veyrunes | <p>Sexually dimorphic behaviours, such as parental care, have long been thought to be driven mostly, if not exclusively, by gonadal hormones. In the past two decades, a few studies have challenged this view, highlighting the direct influence of th... | | Behavior & Social Evolution, Evolutionary Ecology, Reproduction and Sex | Gabriel Marais | 2022-04-08 20:09:58 | ||
24 Oct 2022

Evolutionary responses of energy metabolism, development, and reproduction to artificial selection for increasing heat tolerance in Drosophila subobscuraThe other side of the evolution of heat tolerance: correlated responses in metabolism and life-history traitsRecommended by Inês Fragata and Pedro Simões based on reviews by Marija Savić Veselinović and 1 anonymous reviewer
Understanding how species respond to environmental changes is becoming increasingly important in order to predict the future of biodiversity and species distributions under current global warming conditions (Rezende 2020; Bennett et al 2021). Two key factors to take into account in these predictions are the tolerance of organisms to heat stress and subsequently how they adapt to increasingly warmer temperatures. Coupled with this, one important factor that is often overlooked when addressing the evolution of thermal tolerance, is the correlated responses in traits that are important to fitness, such as life histories, behavior and the underlying metabolic processes. The rate and intensity of the thermal stress are expected to be major factors in shaping the evolution of heat tolerance and correlated responses in other traits. For instance, lower rates of thermal stress are predicted to select for individuals with a slower metabolism (Santos et al 2012), whereas low metabolism is expected to lead to a lower reproductive rate (Dammhahn et al 2018). To quantify the importance of the rate and intensity of thermal stress on the evolutionary response of heat tolerance and correlated response in behavior, Mesas et al (2021) performed experimental evolution in Drosophila subobscura using selective regimes with slow or fast ramping protocols. Whereas both regimes showed increased heat tolerance with similar evolutionary rates, the correlated responses in thermal performance curves for locomotor behavior differed between selection regimes. These findings suggest that thermal rate and intensity may shape the evolution of correlated responses in other traits, urging the need to understand possible correlated responses at relevant levels such as life history and metabolism. In the present contribution, Mesas and Castañeda (2022) investigate whether the disparity in thermal performance curves observed in the previous experiment (Mesas et al 2021) could be explained by differences in metabolic energy production and consumption, and how this correlated with the reproductive output (fecundity and viability). Overall, the authors show some evidence for lowered enzyme activity and increased performance in life-history traits, particularly for the slow-ramping selected flies. Specifically, the authors observe a reduction in glucose metabolism and increased viability when evolving under slow ramping stress. Interestingly, both regimes show a general increase in fecundity, suggesting that adaptation to these higher temperatures is not costly (for reproduction) in the ancestral environment. The evidence for a somewhat lower metabolism in the slow-ramping lines suggests the evolution of a slow “pace of life”. The “pace of life” concept tries to bridge variation across several levels namely metabolism, physiology, behavior and life history, with low “pace of life” organisms presenting lower metabolic rates, later reproduction and higher longevity than fast “pace of life” organisms (Dammhahn et al 2018, Tuzun & Stocks 2022). As the authors state there is not a clear-cut association with the expectations of the pace of life hypothesis since there was evidence for increased reproductive output under both selection intensity regimes. This suggests that, given sufficient trait genetic variance, positively correlated responses may emerge during some stages of thermal evolution. As fecundity estimates in this study were focussed on early life, the possibility of a decrease in the cumulative reproductive output of the selected flies, even under benign conditions, cannot be excluded. This would help explain the apparent paradox of increased fecundity in selected lines. In this context, it would also be interesting to explore the variation in reproductive output at different temperatures, i.e to obtain thermal performance curves for life histories. Mesas and Castañeda (2022) raise important questions to pursue in the future and contribute to the growing evidence that, in order to predict the distribution of ectothermic species under current global warming conditions, we need to expand beyond determining the physiological thermal limits of each organism (Parratt et al 2021). Ultimately, integrating metabolic, life-history and behavioral changes during evolution under different thermal stresses within a coherent framework is key to developing better predictions of temperature effects on natural populations. Bennett, J.M., Sunday, J., Calosi, P. et al. The evolution of critical thermal limits of life on Earth. Nat Commun 12, 1198 (2021). https://doi.org/10.1038/s41467-021-21263-8 Dammhahn, M., Dingemanse, N.J., Niemelä, P.T. et al. Pace-of-life syndromes: a framework for the adaptive integration of behaviour, physiology and life history. Behav Ecol Sociobiol 72, 62 (2018). https://doi.org/10.1007/s00265-018-2473-y Mesas, A, Jaramillo, A, Castañeda, LE. Experimental evolution on heat tolerance and thermal performance curves under contrasting thermal selection in Drosophila subobscura. J Evol Biol 34, 767– 778 (2021). https://doi.org/10.1111/jeb.13777 Parratt, S.R., Walsh, B.S., Metelmann, S. et al. Temperatures that sterilize males better match global species distributions than lethal temperatures. Nat. Clim. Chang. 11, 481–484 (2021). https://doi.org/10.1038/s41558-021-01047-0 Santos, M, Castañeda, LE, Rezende, EL Keeping pace with climate change: what is wrong with the evolutionary potential of upper thermal limits? Ecology and evolution, 2(11), 2866-2880 (2012). https://doi.org/10.1002/ece3.385 Tüzün, N, Stoks, R. A fast pace-of-life is traded off against a high thermal performance. Proceedings of the Royal Society B, 289(1972), 20212414 (2022). https://doi.org/10.1098/rspb.2021.2414 Rezende, EL, Bozinovic, F, Szilágyi, A, Santos, M. Predicting temperature mortality and selection in natural Drosophila populations. Science, 369(6508), 1242-1245 (2020). https://doi.org/10.1126/science.aba9287 | Evolutionary responses of energy metabolism, development, and reproduction to artificial selection for increasing heat tolerance in Drosophila subobscura | Andres Mesas, Luis E. Castaneda | <p>Adaptations to warming conditions exhibited by ectotherms include increasing heat tolerance but also metabolic changes to reduce maintenance costs (metabolic depression), which can allow them to redistribute the energy surplus to biological fun... | | Adaptation, Evolutionary Ecology, Experimental Evolution, Life History | Inês Fragata | 2022-02-08 01:05:50 | ||
11 Oct 2022
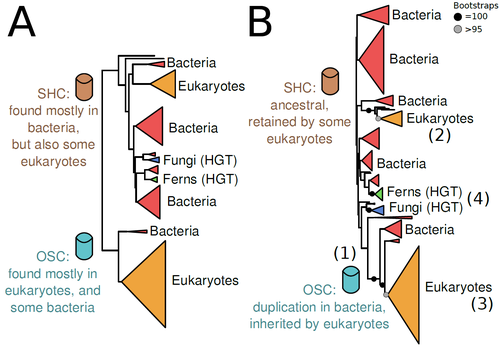
The Eukaryotic Last Common Ancestor Was Bifunctional for Hopanoid and Sterol ProductionGene family analysis suggests new evolutionary scenario for sterol and hopanoid biomarkersRecommended by Iker Irisarri based on reviews by Samuel Abalde, Denis Baurain and Jose Ramon Pardos-BlasSterols and hopanoids are sometimes used as biomarkers to infer the origin of certain groups of organisms. Traditionally, hopanoid-derived products in ancient rocks have been considered to indicate the presence of bacteria, whereas sterol derivatives have been considered to be exclusive to eukaryotes. However, a closer look at the topic reveals a rather complex distribution of either compound in both bacteria and eukaryotes. (1). The known biosynthetic pathways for sterols and hopanoids are similar but diverge at a critical step where two different enzymes are used: squalene-hopene cyclase (SHC) and oxidosqualene cyclase (OSC), the latter requiring oxygen. These two enzymes belong to the same gene family, whose complex evolutionary history is difficult to reconcile with the known species phylogeny. In this study (2), Dr. Warren R. Francis revisits the evolution of this gene family using an extended dataset with a broader taxonomic representation. In contrast to the traditional representation of the tree rooted between SHC and OSC paralogs (i.e., based on function), the author proposes that rooting the tree within bacterial SHCs and assuming a secondary origin of OSC is more parsimonious. This postulates SHC to be the ancestral function –retained in many extant bacteria and some eukaryotes– and OSC to have emerged later within bacteria –currently being mostly present in eukaryotes–. The reconstructed evolutionary history is arguably complex and can only be reconciled with the species' phylogeny by invoking many secondary losses. These losses are considered likely because many extant species acquire sterols and hopanoids by diet and lack one or both enzymes. Some cases of recent horizontal gene transfer are also proposed. In contrast to the dichotomy between bacterial SHCs and eukaryote OSCs, the new proposed scenario suggests that the eukaryote ancestor likely inherited both enzymes from bacteria and thus could be able to synthesize both sterols and hopanoids. Under this hypothesis, not only bacteria but also eukaryotes could be responsible for the hopane found in old rocks. This agrees with eukaryote fossils dating back to more than 1 billion years ago (3). Also, the observed increase of sterane levels in rocks ~600-700 million years old cannot be associated with the origin of eukaryotes, which is a much older event, but could rather reflect changes in atmospheric oxygen levels because oxygen is required for the synthesis of sterols by OSC. References 1. Santana-Molina C, Rivas-Marin E, Rojas AM, Devos DP (2020) Origin and Evolution of Polycyclic Triterpene Synthesis. Molecular Biology and Evolution, 37, 1925–1941. https://doi.org/10.1093/molbev/msaa054 2. Francis WR (2022) The Eukaryotic Last Common Ancestor Was Bifunctional for Hopanoid and Sterol Production. Preprints, 2020040186, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.20944/preprints202004.0186.v5 3. Butterfield NJ (2000) Bangiomorpha pubescens n. gen., n. sp.: implications for the evolution of sex, multicellularity, and the Mesoproterozoic/Neoproterozoic radiation of eukaryotes. Paleobiology, 26, 386–404. https://doi.org/10.1666/0094-8373(2000)026<0386:BPNGNS>2.0.CO;2 | The Eukaryotic Last Common Ancestor Was Bifunctional for Hopanoid and Sterol Production | Warren R Francis | <p>Steroid and hopanoid biomarkers can be found in ancient rocks and may give a glimpse of what life was present at that time. Sterols and hopanoids are produced by two related enzymes, though the evolutionary history of this protein family is com... | | Bioinformatics & Computational Biology, Evolutionary Ecology, Molecular Evolution, Paleontology, Phylogenetics / Phylogenomics | Iker Irisarri | 2021-01-13 16:03:29 | ||
06 Oct 2022
Evolution of sperm morphology in a crustacean genus with fertilization inside an open brood pouch.Evolution of sperm morphology in Daphnia within a phyologenetic contextRecommended by Ellen Decaestecker based on reviews by Renate Matzke-Karasz and 1 anonymous reviewerIn this study sperm morphology is studied in 15 Daphnia species and the morphological data are mapped on a Daphnia phylogeny. The authors found that despite the internal fertilization mode, Daphnia have among the smallest sperm recorded, as would be expected with external fertilization. The authors also conclude that increase in sperm length has evolved twice, that sperm encapsulation has been lost in a clade, and that this clade has very polymorphic sperm with long, and often numerous, filopodia. Daphnia is an interesting model to study sperm morphology because the biology of sexual reproduction is often ignored in (cyclical) parthenogenetic species. Daphnia is part of the very diverse and successful group of cladocerans with cyclical parthenogenetic reproduction. The success of this reproduction mode is reflected in the known 620 species that radiated within this order, this is more than half of the known Branchiopod species diversity and the estimated number of cladoceran species is even two to four times higher (Forró et al. 2008). Looking at this particular model with a good phylogeny and some particularity in the mode of fertilization/reproduction, has thus a large value. Most Daphnia species are cyclical parthenogenetic and switch between sexual and asexual reproduction depending on the environmental conditions. Within the genus Daphnia, evolution to obligate asexuality has evolved in at least four independent occasions by three different mechanisms: (i) obligate parthenogenesis through hybridisation with or without polyploidy, (ii) asexuality has been acquired de novo in some populations and (iii) in certain lineages females reproduce by obligate parthenogenesis, whereas the clonally propagated males produce functional haploid sperm that allows them to breed with sexual females of normal cyclically parthenogenetic lineages (more on this in Decaestecker et al. 2009). This study is made in the context of a body of research on the evolution of one of the most fundamental and taxonomically diverse cell types. There is surprisingly little known about the adaptive value underlying their morphology because it is very difficult to test this experimentally. Studying sperm morphology across species is interesting to study evolution itself because it is a "simple trait". As the authors state: The understanding of the adaptive value of sperm morphology, such as length and shape, remains largely incomplete (Lüpold & Pitnick, 2018). Based on phylogenetic analyses across the animal kingdom, the general rule seems to be that fertilization mode (i.e. whether eggs are fertilized within or outside the female) is a key predictor of sperm length (Kahrl et al., 2021). There is a trade-off between sperm number and length (Immler et al., 2011). This study reports on one of the smallest sperm recorded despite the fertilization being internal. The brood pouch in Daphnia is an interesting particularity as fertilisation occurs internally, but it is not disconnected from the environment. It is also remarkable that there are two independent evolution lines of sperm size in this group. It suggests that those traits have an adaptive value. References Decaestecker E, De Meester L, Mergeay J (2009) Cyclical Parthenogenesis in Daphnia: Sexual Versus Asexual Reproduction. In: Lost Sex: The Evolutionary Biology of Parthenogenesis (eds Schön I, Martens K, Dijk P), pp. 295–316. Springer Netherlands, Dordrecht. https://doi.org/10.1007/978-90-481-2770-2_15 Duneau David, Möst M, Ebert D (2022) Evolution of sperm morphology in a crustacean genus with fertilization inside an open brood pouch. bioRxiv, 2020.01.31.929414, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.01.31.929414 Forró L, Korovchinsky NM, Kotov AA, Petrusek A (2008) Global diversity of cladocerans (Cladocera; Crustacea) in freshwater. Hydrobiologia, 595, 177–184. https://doi.org/10.1007/s10750-007-9013-5 Immler S, Pitnick S, Parker GA, Durrant KL, Lüpold S, Calhim S, Birkhead TR (2011) Resolving variation in the reproductive tradeoff between sperm size and number. Proceedings of the National Academy of Sciences, 108, 5325–5330. https://doi.org/10.1073/pnas.1009059108 Kahrl AF, Snook RR, Fitzpatrick JL (2021) Fertilization mode drives sperm length evolution across the animal tree of life. Nature Ecology & Evolution, 5, 1153–1164. https://doi.org/10.1038/s41559-021-01488-y Lüpold S, Pitnick S (2018) Sperm form and function: what do we know about the role of sexual selection? Reproduction, 155, R229–R243. https://doi.org/10.1530/REP-17-0536 | Evolution of sperm morphology in a crustacean genus with fertilization inside an open brood pouch. | Duneau, David; Moest, Markus; Ebert, Dieter | <p style="text-align: justify;">Sperm is the most fundamental male reproductive feature. It serves the fertilization of eggs and evolves under sexual selection. Two components of sperm are of particular interest, their number and their morphology.... | | Evolutionary Ecology, Morphological Evolution, Reproduction and Sex, Sexual Selection | Ellen Decaestecker | 2020-05-30 22:54:15 | ||
05 Oct 2022
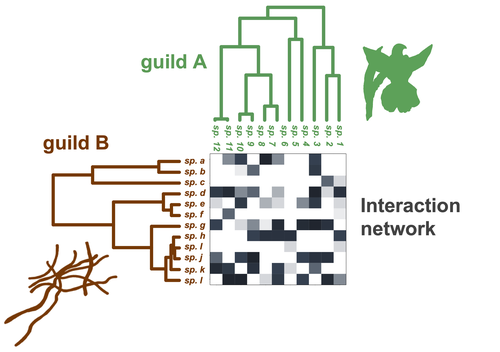
Do closely related species interact with similar partners? Testing for phylogenetic signal in bipartite interaction networksTesting for phylogenetic signal in species interaction networksRecommended by Alejandro Gonzalez-Voyer based on reviews by Joaquin Calatayud and Thomas Guillerme
Species are immersed within communities in which they interact mutualistically, as in pollination or seed dispersal, or nonreciprocally, such as in predation or parasitism, with other species and these interactions play a paramount role in shaping biodiversity (Bascompte and Jordano 2013). Researchers have become increasingly interested in the processes that shape these interactions and how these influence community structure and responses to disturbances. Species interactions are often described using bipartite interaction networks and one important question is how the evolutionary history of the species involved influences the network, including whether there is phylogenetic signal in interactions, in other words whether closely related species interact with other closely related species (Bascompte and Jordano 2013, Perez-Lamarque et al. 2022). To address this question different approaches, correlative and model-based, have been developed to test for phylogenetic signal in interactions, although comparative analyses of the performance of these different metrics are lacking. In their article Perez-Lamarque et al. (2022) set out to test the statistical performance of two widely-used methods, Mantel tests and Phylogenetic Bipartite Linear Models (PBLM; Ives and Godfray 2006) using simulations. Phylogenetic signal is measured as the degree to which distance to the nearest common ancestor predicts the observed similarity in trait values among species. In species interaction networks, the data are actually the between-species dissimilarity among interacting species (Perez-Lamarque et al. 2022), and typical approaches to test for phylogenetic signal cannot be used. However, the Mantel test provides a useful means of analyzing the correlation between two distance matrices, the between-species phylogenetic distance and the between-species dissimilarity in interactions. The PBLM approach, on the other hand, assumes that interactions between species are influenced by unobserved traits that evolve along the phylogenies following a given phenotypic evolution model and the parameters of this model are interpreted in terms of phylogenetic signal (Ives and Godfray 2006). Perez-Lamarque et al (2022) found that the model-based PBLM approach has a high type-I error rate, in other words it often detected phylogenetic signal when there was none. The simple Mantel test was found to present a low type-I error rate and moderate statistical power. However, it tended to overestimate the degree to which species interact with dissimilar partners. In addition to the aforementioned analyses, the authors also tested whether the simple Mantel test was able to detect phylogenetic signal in interactions among species within a given clade in the phylogeny, as phylogenetic signal in species interactions may be localized within specific clades. The article concludes with general guidelines for users wishing to test phylogenetic signal in their interaction networks and illustrates them with an example of an orchid-mycorrhizal fungus network from the oceanic island of La Réunion (Martos et al 2012). This broadly accessible article provides a valuable analysis of the performance of tests of phylogenetic signal in interaction networks enabling users to make informed choices of the analytical methods they wish to employ, and provide useful and detailed guidelines. Therefore, the work should be of broad interest to researchers studying species interactions. References Bascompte J, Jordano P (2013) Mutualistic Networks. Princeton University Press. https://doi.org/10.1515/9781400848720 Ives AR, Godfray HCJ (2006) Phylogenetic Analysis of Trophic Associations. The American Naturalist, 168, E1–E14. https://doi.org/10.1086/505157 Martos F, Munoz F, Pailler T, Kottke I, Gonneau C, Selosse M-A (2012) The role of epiphytism in architecture and evolutionary constraint within mycorrhizal networks of tropical orchids. Molecular Ecology, 21, 5098–5109. https://doi.org/10.1111/j.1365-294X.2012.05692.x Perez-Lamarque B, Maliet O, Pichon B, Selosse M-A, Martos F, Morlon H (2022) Do closely related species interact with similar partners? Testing for phylogenetic signal in bipartite interaction networks. bioRxiv, 2021.08.30.458192, ver. 6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.08.30.458192 | Do closely related species interact with similar partners? Testing for phylogenetic signal in bipartite interaction networks | Benoît Perez-Lamarque, Odile Maliet, Benoît Pichon, Marc-André Selosse, Florent Martos, Hélène Morlon | <p style="text-align: justify;">Whether interactions between species are conserved on evolutionary time-scales has spurred the development of both correlative and process-based approaches for testing phylogenetic signal in interspecific interactio... | | Evolutionary Ecology, Species interactions | Alejandro Gonzalez-Voyer | 2022-03-10 13:48:15 | ||
29 Sep 2022
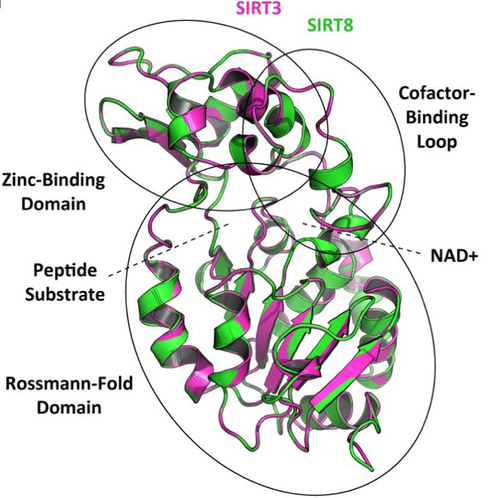
How many sirtuin genes are out there? evolution of sirtuin genes in vertebrates with a description of a new family memberMaking sense of vertebrate sirtuin genesRecommended by Frédéric Delsuc based on reviews by Filipe Castro, Nicolas Leurs and 1 anonymous reviewerSirtuin proteins are class III histone deacetylases that are involved in a variety of fundamental biological functions mostly related to aging. These proteins are located in different subcellular compartments and are associated with different biological functions such as metabolic regulation, stress response, and cell cycle control [1]. In mammals, the sirtuin gene family is composed of seven paralogs (SIRT1-7) grouped into four classes [2]. Due to their involvement in maintaining cell cycle integrity, sirtuins have been studied as a way to understand fundamental mechanisms governing longevity [1]. Indeed, the downregulation of sirtuin genes with aging seems to explain much of the pathophysiology that accumulates with aging [3]. Biomedical studies have thus explored the potential therapeutic implications of sirtuins [4] but whether they can effectively be used as molecular targets for the treatment of human diseases remains to be demonstrated [1]. Despite this biomedical interest and some phylogenetic analyses of sirtuin paralogs mostly conducted in mammals, a comprehensive evolutionary analysis of the sirtuin gene family at the scale of vertebrates was still lacking. In this preprint, Opazo and collaborators [5] took advantage of the increasing availability of whole-genome sequences for species representing all main groups of vertebrates to unravel the evolution of the sirtuin gene family. To do so, they undertook a phylogenomic approach in its original sense aimed at improving functional predictions by evolutionary analysis [6] in order to inventory the full vertebrate sirtuin gene repertoire and reconstruct its precise duplication history. Harvesting genomic databases, they extracted all predicted sirtuin proteins and performed phylogenetic analyses based on probabilistic inference methods. Maximum likelihood and Bayesian analyses resulted in well-resolved and congruent phylogenetic trees dividing vertebrate sirtuin genes into three major clades. These analyses also revealed an additional eighth paralog that was previously overlooked because of its restricted phyletic distribution. This newly identified sirtuin family member (named SIRT8) was recovered with unambiguous statistical support as a sister-group to the SIRT3 clade. Comparative genomic analyses based on conserved gene synteny confirmed that SIRT8 was present in all sampled non-amniote vertebrate genomes (cartilaginous fish, bony fish, coelacanth, lungfish, and amphibians) except cyclostomes. SIRT8 has thus most likely been lost in the last common ancestor of amniotes (mammals, reptiles, and birds). Discovery of such previously unknown genes in vertebrates is not completely surprising given the plethora of high-quality genomes now available. However, this study highlights the importance of considering a broad taxonomic sampling to infer evolutionary patterns of gene families that have been mostly studied in mammals because of their potential importance for human biology. Based on its phylogenetic position as closely related to SIRT3 within class I, it could be predicted that the newly identified SIRT8 paralog likely has a deacetylase activity and is probably located in mitochondria. To test these evolutionary predictions, Opazo and collaborators [5] conducted further bioinformatics analyses and functional experiments using the elephant shark (Callorhinchus milii) as a model species. RNAseq expression data were analyzed to determine tissue-specific transcription of sirtuin genes in vertebrates, including SIRT8 found to be mainly expressed in the ovary, which suggests a potential role in biological processes associated with reproduction. The elephant shark SIRT8 protein sequence was used with other vertebrates for comparative analyses of protein structure modeling and subcellular localization prediction both pointing to a probable mitochondrial localization. The protein localization and its function were further characterized by immunolocalization in transfected cells, and enzymatic and functional assays, which all confirmed the prediction that SIRT8 proteins are targeted to the mitochondria and have deacetylase activity. The extensive experimental efforts made in this study to shed light on the function of this newly discovered gene are both rare and highly commendable. Overall, this work by Opazo and collaborators [5] provides a comprehensive phylogenomic study of the sirtuin gene family in vertebrates based on detailed evolutionary analyses using state-of-the-art phylogenetic reconstruction methods. It also illustrates the power of adopting an integrative comparative approach supplementing the reconstruction of the duplication history of the gene family with complementary functional experiments in order to elucidate the function of the newly discovered SIRT8 family member. These results provide a reference phylogenetic framework for the evolution of sirtuin genes and the further functional characterization of the eight vertebrate paralogs with potential relevance for understanding the cellular biology of aging and its associated diseases in human. References [1] Vassilopoulos A, Fritz KS, Petersen DR, Gius D (2011) The human sirtuin family: Evolutionary divergences and functions. Human Genomics, 5, 485. https://doi.org/10.1186/1479-7364-5-5-485 [2] Yamamoto H, Schoonjans K, Auwerx J (2007) Sirtuin Functions in Health and Disease. Molecular Endocrinology, 21, 1745–1755. https://doi.org/10.1210/me.2007-0079 [3] Morris BJ (2013) Seven sirtuins for seven deadly diseases ofaging. Free Radical Biology and Medicine, 56, 133–171. https://doi.org/10.1016/j.freeradbiomed.2012.10.525 [4] Bordo D Structure and Evolution of Human Sirtuins. Current Drug Targets, 14, 662–665. http://dx.doi.org/10.2174/1389450111314060007 [5] Opazo JC, Vandewege MW, Hoffmann FG, Zavala K, Meléndez C, Luchsinger C, Cavieres VA, Vargas-Chacoff L, Morera FJ, Burgos PV, Tapia-Rojas C, Mardones GA (2022) How many sirtuin genes are out there? evolution of sirtuin genes in vertebrates with a description of a new family member. bioRxiv, 2020.07.17.209510, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.07.17.209510 [6] Eisen JA (1998) Phylogenomics: Improving Functional Predictions for Uncharacterized Genes by Evolutionary Analysis. Genome Research, 8, 163–167. https://doi.org/10.1101/gr.8.3.163 | How many sirtuin genes are out there? evolution of sirtuin genes in vertebrates with a description of a new family member | Juan C. Opazo, Michael W. Vandewege, Federico G. Hoffmann, Kattina Zavala, Catalina Meléndez, Charlotte Luchsinger, Viviana A. Cavieres, Luis Vargas-Chacoff, Francisco J. Morera, Patricia V. Burgos, Cheril Tapia-Rojas, Gonzalo A. Mardones | <p style="text-align: justify;">Studying the evolutionary history of gene families is a challenging and exciting task with a wide range of implications. In addition to exploring fundamental questions about the origin and evolution of genes, disent... | | Molecular Evolution | Frédéric Delsuc | Filipe Castro, Anonymous, Nicolas Leurs | 2022-05-12 16:06:04 | |
15 Sep 2022

Bimodal breeding phenology in the Parsley Frog Pelodytes punctatus as a bet-hedging strategy in an unpredictable environment despite strong priority effectsSpreading the risk of reproductive failure when the environment is unpredictable and ephemeralRecommended by Gabriele Sorci based on reviews by Thomas Haaland and Zoltan RadaiMany species breed in environments that are unpredictable, for instance in terms of the availability of resources needed to raise the offspring. Organisms might respond to such spatial and temporal unpredictability by adopting plastic responses to adjust their reproductive investment according to perceived cues of environmental quality. Some species such as the amphibians might also face the problem of ephemeral habitats, when the ponds where they breed have a chance of drying up before metamorphosis has occurred. In this case, maximizing long-term fitness might involve a strategy of spreading the risk, even though the reproductive success of a single reproductive bout might be lower. Understanding how animals (and plants) get adapted to stochastic environments is particularly crucial in the current context of rapid environmental changes. In this article, Jourdan-Pineau et al. report the results of field surveys of the Parsley Frog (Pelodytes punctatus) in Southern France. This frog has peculiar breeding phenology with females breeding in autumn and spring. The authors provide quite an extensive amount of information on the reproductive success of eggs laid in each season and the possible ecological factors accounting for differences between seasons. Although the presence of interspecific competitors and predators does not seem to account for pond-specific reproductive success, the survival of tadpoles hatching from eggs laid in spring is severely impaired when tadpoles from the autumn cohort have managed to survive. This intraspecific competition takes the form of a “priority” effect where tadpoles from the autumn cohort outcompete the smaller larvae from the spring cohort. Given this strong priority effect, one might tentatively predict that females laying in spring should avoid ponds with tadpoles from the autumn cohort. Surprisingly, however, the authors did not find any evidence for such avoidance, which might indicate strong constraints on the availability of ponds where females might possibly lay. Assuming that each female can indeed lay both in autumn and spring, how is this bimodal phenology maintained? Would not be worthier to allocate all the eggs to the autumn (or the spring) laying season? Eggs laid in autumn and spring have to face different environmental hazards, reducing their hatching success and the probability to produce metamorphs (for instance, tadpoles hatching from eggs laid in autumn have to overwinter which might be a particularly risky phase). Jourdan-Pineau and coworkers addressed this question by adapting a bet-hedging model that was initially developed to investigate the strategy of allocation into seed dormancy of annual plants (Cohen 1966) to the case of the bimodal phenology of the Parsley Frog. By feeding the model with the parameter values obtained from the field surveys, they found that the two breeding strategies (laying in autumn and in spring) can coexist as long as the probability of breeding success in the autumn cohort is between 20% and 80% (the range of values allowing the coexistence of a bimodal phenology shrinking a little bit when considering that frogs can reproduce 5 times during their lifespan instead of three times). This paper provides a very nice illustration of the importance of combining approaches (here field monitoring to gather data that can be used to feed models) to understand the evolution of peculiar breeding strategies. Although future work should attempt to gather individual-based data (in addition to population data), this work shows that spreading the risk can be an adaptive strategy in environments characterized by strong stochastic variation. References Cohen D (1966) Optimizing reproduction in a randomly varying environment. Journal of Theoretical Biology, 12, 119–129. https://doi.org/10.1016/0022-5193(66)90188-3 Jourdan-Pineau H., Crochet P.-A., David P. (2022) Bimodal breeding phenology in the Parsley Frog Pelodytes punctatus as a bet-hedging strategy in an unpredictable environment despite strong priority effects. bioRxiv, 2022.02.24.481784, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.02.24.481784 | Bimodal breeding phenology in the Parsley Frog Pelodytes punctatus as a bet-hedging strategy in an unpredictable environment despite strong priority effects | Helene Jourdan-Pineau, Pierre-Andre Crochet, Patrice David | <p style="text-align: justify;">When environmental conditions are unpredictable, expressing alternative phenotypes spreads the risk of failure, a mixed strategy called bet-hedging. In the southern part of its range, the Parsley Frog <em>Pelodytes ... | | Adaptation, Evolutionary Ecology, Life History | Gabriele Sorci | 2022-02-28 11:53:00 | ||
06 Sep 2022

Masculinization of the X-chromosome in aphid soma and gonadsSex-biased gene expression is not tissue-specific in Pea AphidsRecommended by Charles Baer and Tanja Schwander based on reviews by Ann Kathrin Huylmans and 1 anonymous reviewerSexual antagonism (SA), wherein the fitness interests of the sexes do not align, is inherent to organisms with two (or more) sexes. SA leads to intra-locus sexual conflict, where an allele that confers higher fitness in one sex reduces fitness in the other [1, 2]. This situation leads to what has been referred to as "gender load", resulting from the segregation of SA alleles in the population. Gender load can be reduced by the evolution of sex-specific (or sex-biased) gene expression. A specific prediction is that gene-duplication can lead to sub- or neo-functionalization, in which case the two duplicates partition the function in the different sexes. The conditions for invasion by a SA allele differ between sex-chromosomes and autosomes, leading to the prediction that (in XY or XO systems) the X should accumulate recessive male-favored alleles and dominant female-favored alleles; similar considerations apply in ZW systems ([3, but see 4]. Aphids present an interesting special case, for several reasons: they have XO sex-determination, and three distinct reproductive morphs (sexual females, parthenogenetic females, and males). Previous theoretical work by the lead author predict that the X should be optimized for male function, which was borne out by whole-animal transcriptome analysis [5]. Here [6], the authors extend that work to investigate “tissue”-specific (heads, legs and gonads), sex-specific gene expression. They argue that, if intra-locus SA is the primary driver of sex-biased gene expression, it should be generally true in all tissues. They set up as an alternative the possibility that sex-biased gene expression could also be driven by dosage compensation. They cite references supporting their argument that "dosage compensation (could be) stronger in the brain", although the underlying motivation for that argument appears to be based on empirical evidence rather than theoretical predictions. At any rate, the results are clear: all tissues investigated show masculinization of the X. Further, X-linked copies of gene duplicates were more frequently male-biased than duplicated autosomal genes or X-linked single-copy genes. To sum up, this is a nice empirical study with clearly interpretable (and interpreted) results, the most obvious of which is the greater sex-biased expression in sexually-dimorphic tissues. Unfortunately, as the authors emphasize, there is no general theory by which SA, variable dosage-compensation, and meiotic sex chromosome inactivation can be integrated in a predictive framework. It is to be hoped that empirical studies such as this one will motivate deeper and more general theoretical investigations. References [1] Rice WR, Chippindale AK (2001) Intersexual ontogenetic conflict. Journal of Evolutionary Biology 14: 685-693. https://doi.org/10.1046/j.1420-9101.2001.00319.x [2] Bonduriansky R, Chenoweth SF (2009) Intralocus sexual conflict. Trends Ecol Evol 24: 280-288. https://doi.org/10.1016/j.tree.2008.12.005 [3] Rice WR. (1984) Sex chromosomes and the evolution of sexual dimorphism. Evolution 38: 735-742. https://doi.org/10.1086/595754 [4] Fry JD (2010) The genomic location of sexually antagonistic variation: some cautionary comments. Evolution 64: 1510-1516. https://doi.org/10.1111%2Fj.1558-5646.2009.00898.x [5] Jaquiéry J, Rispe C, Roze D, Legeai F, Le Trionnaire G, Stoeckel S, et al. (2013) Masculinization of the X Chromosome in the Pea Aphid. PLoS Genetics 9. https://doi.org/10.1371/journal.pgen.1003690 [6] Jaquiéry J, Simon J-C, Robin S, Richard G, Peccoud J, Boulain H, Legeai F, Tanguy S, Prunier-Leterme N, Le Trionnaire G (2022) Masculinization of the X-chromosome in aphid soma and gonads. bioRxiv, 2021.08.13.453080, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.08.13.453080 | Masculinization of the X-chromosome in aphid soma and gonads | Julie Jaquiery, Jean-Christophe Simon, Stephanie Robin, Gautier Richard, Jean Peccoud, Helene Boulain, Fabrice Legeai, Sylvie Tanguy, Nathalie Prunier-Leterme, Gael Letrionnaire | <p>Males and females share essentially the same genome but differ in their optimal values for many phenotypic traits, which can result in intra-locus conflict between the sexes. Aphids display XX/X0 sex chromosomes and combine unusual X chromosome... | | Genetic conflicts, Genome Evolution, Reproduction and Sex | Charles Baer | 2021-08-16 08:56:08 | ||
02 Sep 2022

Introgression between highly divergent sea squirt genomes: an adaptive breakthrough?A match made in the Anthropocene: human-mediated adaptive introgression across a speciation continuumRecommended by Fernando Racimo based on reviews by Michael Westbury, Andrew Foote and Erin CalfeeThe long-distance transport and introduction of new species by humans is increasingly leading divergent lineages to interact, and sometimes interbreed, even after thousands or millions of years of separation. It is thus of prime importance to understand the consequences of these contemporary admixture events on the evolutionary fitness of interacting organisms, and their ecological implications. Ciona robusta and Ciona intestinalis are two species of sea squirts that diverged between 1.5 and 2 million years ago and recently came into contact again. This occurred through human-mediated introduction of C. robusta (native to the Northwest Pacific) into the range of C. intestinalis (the English channeled Northeast Atlantic). In this study, Fraïsse et al. (2022) follow up on earlier work by Le Moan et al. (2021), who had identified a long genomic hotspot of introgression of C. robusta ancestry segments in chromosome 5 of C. intestinalis. The hotspot bears suggestive evidence of positive selection and the authors aimed to investigate this further using fully phased whole-genome sequences. The authors narrow down on the exact boundaries of the introgressed region, and make a compelling case that it has been the likely target of positive selection after introgression, using various complementary approaches based on patterns of population differentiation, haplotype structure and local levels of diversity in the region. Using extensive demographic modeling, they also show that the introgression event was likely recent (approximately 75 years ago), and distinct from other tracts in the C. intestinalis genome that are likely a product of more ancient episodes of interbreeding in the past 30,000 years. Narrowing down on the potential drivers of selection, the authors show that candidate SNPs in the region overlap with the cytochrome family 2 subfamily U gene - involved in the detoxification of exogenous compounds - potentially reflecting adaptation to chemicals encountered in the sea squirt's environment. There also appears to be copy number variation at the candidate SNPs, which provides clues into the adaptation mechanism in the region. All reviewers agreed that the work carried out by the authors is elegant and the results are robustly supported and well presented. In a round of reviews, various clarifications of the manuscript were suggested by the reviewers, including on the quality of the newly generated sequencing data, and some suggestions for qualifications on the conclusions reached by the authors as well as changes in the figures to increase their clarity. The authors addressed the different concerns of the reviewers, and the new version is much improved. This study into human-mediated introgression and its consequences for adaptation is, in my view, both well thought-out and executed. I therefore provide an enthusiastic recommendation of this manuscript. References Fraïsse C, Le Moan A, Roux C, Dubois G, Daguin-Thiébaut C, Gagnaire P-A, Viard F and Bierne N (2022) Introgression between highly divergent sea squirt genomes: an adaptive breakthrough? bioRxiv, 2022.03.22.485319, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.03.22.485319 Le Moan A, Roby C, Fraïsse C, Daguin-Thiébaut C, Bierne N, Viard F (2021) An introgression breakthrough left by an anthropogenic contact between two ascidians. Molecular Ecology, 30, 6718–6732. https://doi.org/10.1111/mec.16189 | Introgression between highly divergent sea squirt genomes: an adaptive breakthrough? | Christelle Fraïsse, Alan Le Moan, Camille Roux, Guillaume Dubois, Claire Daguin-Thiébaut, Pierre-Alexandre Gagnaire, Frédérique Viard, Nicolas Bierne | <p style="text-align: justify;">Human-mediated introductions are reshuffling species distribution on a global scale. Consequently, an increasing number of allopatric taxa are now brought into contact, promoting introgressive hybridization between ... | | Adaptation, Hybridization / Introgression, Population Genetics / Genomics | Fernando Racimo | 2022-04-14 15:30:42 |




