
Latest recommendations
| Id | Title * | Authors * | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
25 Sep 2023
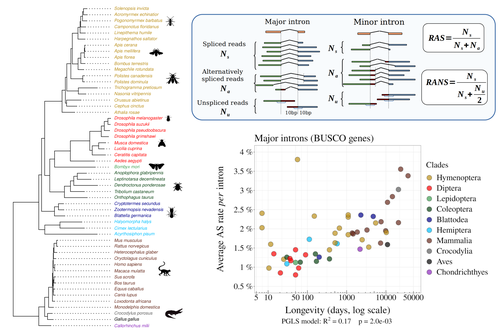
Random genetic drift sets an upper limit on mRNA splicing accuracy in metazoansThe drift barrier hypothesis and the limits to alternative splicing accuracyRecommended by Ignacio Bravo based on reviews by Lars M. Jakt and 2 anonymous reviewers based on reviews by Lars M. Jakt and 2 anonymous reviewers
Accurate information flow is central to living systems. The continuity of genomes through generations as well as the reproducible functioning and survival of the individual organisms require a faithful information transfer during replication, transcription and translation. The differential efficiency of natural selection against “mistakes” results in decreasing fidelity rates for replication, transcription and translation. At each level in the information flow chain (replication, transcription, translation), numerous complex molecular systems have evolved and been selected for preventing, identifying and, when possible, correcting or removing such “mistakes” arising during information transfer. However, fidelity cannot be improved ad infinitum. First, because of the limits imposed by the physical nature of the processes of copying and recoding information over different molecular supports: all mechanisms ensuring fidelity during biological information transfer ultimately rely on chemical kinetics and thermodynamics. The more accurate a copying process is, the lower the synthesis rate and the higher the energetic cost of correcting errors. Second, because of the limits imposed by random genetic drift: natural selection cannot effectively act on an allele that contributes with a small differential advantage unless effective population size is large. If s <1/Ne (or s <1/(2Ne) in diploids) the allele frequency in the population is de facto subject to neutral drift processes. In their preprint “Random genetic drift sets an upper limit on mRNA splicing accuracy in metazoans”, Bénitière, Necsulea and Duret explore the validity of this last mentioned “drift barrier” hypothesis for the case study of alternative splicing diversity in eukaryotes (Bénitière et al. 2022). Splicing refers to an ensemble of eukaryotic molecular processes mediated by a large number of proteins and ribonucleoproteins and involving nucleotide sequence recognition, that uses as a molecular substrate a precursor messenger RNA (mRNA), directly transcribed from the DNA, and produces a mature mRNA by removing introns and joining exons (Chow et al. 1977). Alternative splicing refers to the case in which different molecular species of mature mRNAs can be produced, either by cis-splicing processes acting on the same precursor mRNA, e.g. by varying the presence/absence of different exons or by varying the exon-exon boundaries, or by trans-splicing processes, joining exons from different precursor mRNA molecules. The diversity of mRNA molecular species generated by alternative splicing enlarges the molecular phenotypic space that can be generated from the same genotype. In humans, alternative splicing occurs in around 95% of the ca. 20,000 genes, resulting in ca. 100,000 medium-to-high abundance transcripts (Pan et al. 2008). In multicellular organisms, the frequency of alternatively spliced mRNAs varies between tissues and across ontogeny, often in a switch-like pattern (Wang et al. 2008). In the molecular and cell biology community, it is commonly accepted that splice variants contribute with specific functions (Marasco and Kornblihtt 2023) although there exists a discussion around the functional nature of low-frequency splice variants (see for instance the debate between Tress et al. 2017 and Blencowe 2017). The origin, diversity, regulation and evolutionary advantage of alternative splicing constitutes thus a playground of the selectionist-neutralist debate, with one extreme considering that most splice variants are mere “mistakes” of the splicing process (Pickrell et al. 2010), and the other extreme considering that alternative splicing is at the core of complexity in multicellular organisms, as it increases the genome coding potential and allows for a large repertoire of cell types (Chen et al. 2014). In their manuscript, Bénitière, Necsulea and Duret set the cursor towards the neutralist end of the gradient and test the hypothesis of whether the high alternative splice rate in “complex” organisms corresponds to a high rate of splicing “mistakes”, arising from the limit imposed by the drift barrier effect on the power of natural selection to increase accuracy (Bush et al. 2017). In their preprint, the authors convincingly show that in metazoans a fraction of the variation of alternative splicing rate is explained by variation in proxies of population size, so that species with smaller Ne display higher alternative splice rates. They communicate further that abundant splice variants tend to preserve the reading frame more often than low-frequency splice variants, and that the nucleotide splice signals in abundant splice variants display stronger evidence of purifying selection than those in low-frequency splice variants. From all the evidence presented in the manuscript, the authors interpret that “variation in alternative splicing rate is entirely driven by variation in the efficacy of selection against splicing errors”. The authors honestly present some of the limitations of the data used for the analyses, regarding i) the quality of the proxies used for Ne (i.e. body length, longevity and dN/dS ratio); ii) the heterogeneous nature of the RNA sequencing datasets (full organisms, organs or tissues; different life stages, sexes or conditions); and iii) mostly short RNA reads that do not fully span individual introns. Further, data from bacteria do not verify the herein communicated trends, as it has been shown that bacterial species with low population sizes do not display higher transcription error rates (Traverse and Ochman 2016). Finally, it will be extremely interesting to introduce a larger evolutionary perspective on alternative splicing rates encompassing unicellular eukaryotes, in which an intriguing interplay between alternative splicing and gene duplication has been communicated (Hurtig et al. 2020). The manuscript from Bénitière, Necsulea and Duret makes a significant advance to our understanding of the diversity, the origin and the physiology of post-transcriptional and post-translational mechanisms by emphasising the fundamental role of non-adaptive evolutionary processes and the upper limits to splicing accuracy set by genetic drift. References Bénitière F, Necsulea A, Duret L. 2023. Random genetic drift sets an upper limit on mRNA splicing accuracy in metazoans. bioRxiv, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.12.09.519597 Blencowe BJ. 2017. The Relationship between Alternative Splicing and Proteomic Complexity. Trends Biochem Sci 42:407–408. https://doi.org/10.1016/j.tibs.2017.04.001 Bush SJ, Chen L, Tovar-Corona JM, Urrutia AO. 2017. Alternative splicing and the evolution of phenotypic novelty. Philos Trans R Soc Lond B Biol Sci 372:20150474. https://doi.org/10.1098/rstb.2015.0474 Chen L, Bush SJ, Tovar-Corona JM, Castillo-Morales A, Urrutia AO. 2014. Correcting for differential transcript coverage reveals a strong relationship between alternative splicing and organism complexity. Mol Biol Evol 31:1402–1413. https://doi.org/10.1093/molbev/msu083 Chow LT, Gelinas RE, Broker TR, Roberts RJ. 1977. An amazing sequence arrangement at the 5’ ends of adenovirus 2 messenger RNA. Cell 12:1–8. https://doi.org/10.1016/0092-8674(77)90180-5 Hurtig JE, Kim M, Orlando-Coronel LJ, Ewan J, Foreman M, Notice L-A, Steiger MA, van Hoof A. 2020. Origin, conservation, and loss of alternative splicing events that diversify the proteome in Saccharomycotina budding yeasts. RNA 26:1464–1480. https://doi.org/10.1261/rna.075655.120 Marasco LE, Kornblihtt AR. 2023. The physiology of alternative splicing. Nat Rev Mol Cell Biol 24:242–254. https://doi.org/10.1038/s41580-022-00545-z Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. 2008. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40:1413–1415. https://doi.org/10.1038/ng.259 Pickrell JK, Pai AA, Gilad Y, Pritchard JK. 2010. Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet 6:e1001236. https://doi.org/10.1371/journal.pgen.1001236 Traverse CC, Ochman H. 2016. Conserved rates and patterns of transcription errors across bacterial growth states and lifestyles. Proc Natl Acad Sci U S A 113:3311–3316. https://doi.org/10.1073/pnas.1525329113 Tress ML, Abascal F, Valencia A. 2017. Alternative Splicing May Not Be the Key to Proteome Complexity. Trends Biochem Sci 42:98–110. https://doi.org/10.1016/j.tibs.2016.08.008 Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456:470–476. https://doi.org/10.1038/nature07509 | Random genetic drift sets an upper limit on mRNA splicing accuracy in metazoans | Florian Benitiere, Anamaria Necsulea, Laurent Duret | <p style="text-align: justify;">Most eukaryotic genes undergo alternative splicing (AS), but the overall functional significance of this process remains a controversial issue. It has been noticed that the complexity of organisms (assayed by the nu... | | Bioinformatics & Computational Biology, Genome Evolution, Molecular Evolution, Population Genetics / Genomics | Ignacio Bravo | Anonymous | 2022-12-12 14:00:01 | |
07 Aug 2023

Pollen-feeding delays reproductive senescence and maintains toxicity of Heliconius eratoImpact of pollen-feeding on egg-laying and cyanogenic glucoside abundance in red postman butterfliesRecommended by Adriana Briscoe based on reviews by Carol Boggs, Caroline Mueller and 1 anonymous reviewerGrowth, development and reproduction in animals are all limited by dietary nutrients. Expansion of an organism’s diet to sources not accessible to closely related species reduces food competition, and eases the constraints of nutrient-limited diets. Adult butterflies are herbivorous insects known to feed primarily on nectar from flowers, which is rich in sugars but poor in amino acids. Only certain species in the genus Heliconius are known to also feed on pollen, which is especially rich in amino acids, and is known to prolong their lives by several months. The ability to digest pollen in Heliconius has been linked to specialized feeding behaviors (Krenn et al. 2009) and extra-oral digestion using enzymes, possibly including duplicated copies of cocoonase (Harpel et al. 2016; Smith et al. 2016 and 2018), a protease used by some moths to digest silk upon eclosion from their cocoons. In this reprint, Pinheiro de Castro and colleagues investigated the impact of artificial and natural diets on egg-laying ability, body weight, and cyanogenic glucoside abundance in adult Heliconius erato butterflies of both sexes. Previous studies (Dunlap-Pianka et al. 1981) in H. charithonia demonstrated that access to dietary pollen led to extended egg-laying ability among adult female butterflies compared to females deprived of pollen, and compared to Dryas iulia females which feed only on nectar. In the current study, Pinheiro de Castro et al. (2023) examine the impact of diet on both young and old H. erato, over a longer period of time than the earlier work, highlighting the importance of extending the time period over which effects are evaluated. In addition to extending egg-laying ability in older females, the authors found that pollen in the diet appeared to maintain older female body weight, presumably because the pollen contained nutrients depleted during egg-laying. The authors then investigated the effects of nutrition on the production of cyanogenic glycoside defenses. Heliconius are aposematic butterflies that sequester cyanide-forming defense chemicals from food plants as larvae or synthesize these compounds de novo. The authors found the abundance of cyanogenic glycosides to be significantly greater in butterflies with access to pollen, but again only in older females. Curiously, field studies of male and female H. charithonia butterflies found that females in the wild collected more pollen than males (Mendoza-Cuenca and Macías-Ordóñez 2005). Taken together, these new findings raise the intriguing possibility that females collect more pollen than males, in part, because pollen has a bigger impact on female survival and reproduction. A small limitation of the study is the use of wing length, rather than body weight, at the zero time point. But the trend is clear in both males and females, and it adds supporting detail to the efficacy of pollen feeding as an unusual strategy for increasing fertility and survival in Heliconius butterflies.
References | Pollen-feeding delays reproductive senescence and maintains toxicity of Heliconius erato | Erika C. Pinheiro de Castro, Josie McPherson, Glennis Jullian, Anniina L. K. Mattila, Søren Bak, Stephen Montgomery, Chris Jiggins | <p>Dietary shifts may act to ease energetic constraints and allow organisms to optimise life-history traits. Heliconius butterflies differ from other nectar-feeders due to their unique ability to digest pollen, which provides a reliable source of ... | | Evolutionary Ecology, Life History | Adriana Briscoe | 2023-02-07 12:59:54 | ||
04 Aug 2023

Sensitive windows for within- and trans-generational plasticity of anti-predator defencesSensitive windows for phenotypic plasticity within and across generations; where empirical results do not meet the theory but open a world of possibilitiesRecommended by Benoit Pujol based on reviews by David Murray-Stoker, Timothée Bonnet and Willem Frankenhuis
It is easy to define phenotypic plasticity as a mechanism by which traits change in response to a modification of the environment. Many complex mechanisms are nevertheless involved with plastic responses, their strength, and stability (e.g., reliability of cues, type of exposure, genetic expression, epigenetics). It is rather intuitive to think that environmental cues perceived at different stages of development will logically drive different phenotypic responses (Fawcett and Frankenhuis 2015). However, it has proven challenging to try and explain, or model how and why different effects are caused by similar cues experienced at different developmental or life stages (Walasek et al. 2022). The impact of these ‘sensitive windows’ on the stability of plastic responses within or across generations remains unclear. In their paper entitled “Sensitive windows for within- and trans-generational plasticity of anti-predator defences”, Tariel-Adam (2023) address this question. In this paper, Tariel et al. acknowledge the current state of the art, i.e., that some traits influenced by the environment at early life stages become fixed later in life (Snell-Rood et al. 2015) and that sensitive windows are therefore more likely to be observed during early stages of development. Constructive exchanges with the reviewers illustrated that Tariel et al. presented a clear picture of the knowledge on sensitive windows from a conceptual and a mechanistic perspective, thereby providing their study with a strong and elegant rationale. Tariel et al. outlined that little is known about the significance of this scenario when it comes to transgenerational plasticity. Theory predicts that exposure late in the life of parents should be more likely to drive transgenerational plasticity because the cue perceived by parents is more likely to be reliable if time between parental exposure and offspring expression is short (McNamara et al. 2016). I would argue that although sensible, this scenario is likely oversimplifying the complexity of evolutionary, ecological, and inheritance mechanisms at play (Danchin et al. 2018). Tariel-Adam et al. (2023) point out in their paper how the absence of experimental results limits our understanding of the evolutionary and adaptive significance of transgenerational plasticity and decided to address this broad question. Tariel-Adam et al. (2023) used the context of predator-prey interactions, which is a powerful framework to evaluate the temporality of predator cues and prey responses within and across generations (Sentis et al. 2018). They conducted a very elegant experiment whereby two generations of freshwater snails Physa acuta were exposed to crayfish predator cues at different developmental windows. They triggered the within-generation phenotypic plastic response of inducible defences (e.g., shell thickness) and identified sensitive windows as to evaluate their role in within-generation phenotypic plasticity versus transgenerational plasticity. They used different linear models, which lead to constructive exchanges with reviewers, and between reviewers, well trained on these approaches, in particular on effect sizes, that improved the paper by pushing the discussion all the way towards a consensus. Tariel-Adam et al. (2023) results showed that the phenotypic plastic response of different traits was associated with different sensitive windows. Although early-life development was confirmed to be a sensitive window, it was far from being the only developmental stage driving within-generation plastic responses of defence traits. This finding contributes to change our views on plasticity because where theoretical models predict early- and late-life sensitive windows, empirical results gathered here present a more continuous opportunity for sensitive windows over the lifetime of freshwater snails. This is likely because multifactorial mechanisms drive the reliability and adaptive significance of predator cues. To me, this paper most original contribution lies probably in the empirical investigation of sensitive windows underlying transgenerational plasticity. Their finding implies mechanistic ties between sensitive windows driving within-generation and transgenerational plasticity for some traits, but they also shed light on the possible independence of these processes. Although one may be disheartened by these findings illustrating the ability of nature to combine complex mechanisms in order to produce somewhat unpredictable scenarios, one can only find that this unlimited range of phenotypic plasticity scenarios is a wonder to investigate because much remains to be understood. As mentioned in the conclusion of the paper, the opportunity for sensitive windows to drive such a range of plastic responses may also be an opportunity for organisms to adapt to a wide range of environmental demands. References Danchin E, A Pocheville, O Rey, B Pujol, and S Blanchet (2019). Epigenetically facilitated mutational assimilation: epigenetics as a hub within the inclusive evolutionary synthesis. Biological Reviews, 94: 259-282. https://doi.org/10.1111/brv.12453 Fawcett TW, and WE Frankenhuis (2015). Adaptive Explanations for Sensitive Windows in Development. Frontiers in Zoology 12, S3. https://doi.org/10.1186/1742-9994-12-S1-S3 McNamara JM, SRX Dall, P Hammerstein, and O Leimar (2016). Detection vs. Selection: Integration of Genetic, Epigenetic and Environmental Cues in Fluctuating Environments. Ecology Letters 19, 1267–1276. https://doi.org/10.1111/ele.12663 Sentis A, R Bertram, N Dardenne, et al. (2018). Evolution without standing genetic variation: change in transgenerational plastic response under persistent predation pressure. Heredity 121, 266–281. https://doi.org/10.1038/s41437-018-0108-8 Snell-Rood EC, EM Swanson, and RL Young (2015). Life History as a Constraint on Plasticity: Developmental Timing Is Correlated with Phenotypic Variation in Birds. Heredity 115, 379–388. https://doi.org/10.1038/hdy.2015.47 Tariel-Adam J, E Luquet, and S Plénet (2023). Sensitive windows for within- and trans-generational plasticity of anti-predator defences. OSF preprints, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.31219/osf.io/mr8hu Walasek N, WE Frankenhuis, and K Panchanathan (2022). An Evolutionary Model of Sensitive Periods When the Reliability of Cues Varies across Ontogeny. Behavioral Ecology 33, 101–114. https://doi.org/10.1093/beheco/arab113 | Sensitive windows for within- and trans-generational plasticity of anti-predator defences | Juliette Tariel-Adam; Émilien Luquet; Sandrine Plénet | <p>Transgenerational plasticity could be an important mechanism for adaptation to variable environments in addition to within-generational plasticity. But its potential for adaptation may be restricted to specific developmental windows that are hi... | | Adaptation, Evolutionary Ecology, Phenotypic Plasticity | Benoit Pujol | 2022-11-14 08:08:27 | ||
30 Jun 2023
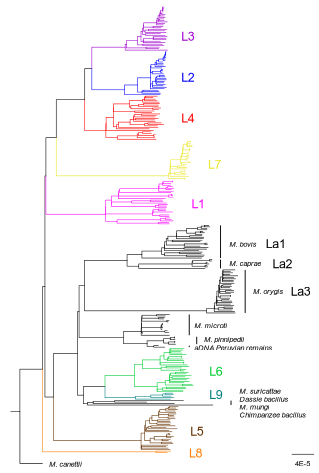
How do monomorphic bacteria evolve? The Mycobacterium tuberculosis complex and the awkward population genetics of extreme clonalityHow the tubercle bacillus got its genome: modernising, modelling, and making sense of the stories we tellRecommended by B. Jesse Shapiro based on reviews by 2 anonymous reviewersIn this instructive review, Stritt and Gagneux offer a balanced perspective on the evolutionary forces shaping Mycobacterium tuberculosis and make the case that our instinct for storytelling be balanced with quantitative models. M. tuberculosis is perhaps the best-known clonal bacterial pathogen – evolving largely in the absence of horizontal gene transfer. Its genome is full of puzzling patterns, including much higher GC content than most intracellular pathogens (which suggests efficient selection to resist AT-skewed mutational bias) but a very high ratio of nonsynonymous to synonymous substitution rates (dN/dS ~ 0.5, typically interpreted as weak selection against deleterious amino acid changes). The authors offer alternative explanations for these patterns, framing the question: is M. tuberculosis evolution shaped mainly by drift or by efficient selection? They propose that this question can only be answered by accounting for the pathogen’s extreme clonality. A clonal lifestyle can have its advantages, for example when adaptations must arise in a particular order (Kondrashov and Kondrashov 2001). An important disadvantage highlighted by the authors are linkage effects: without recombination to shuffle them up, beneficial mutations are linked to deleterious mutations in the same genome (hitchhiking) and purging deleterious mutations also purges neutral diversity across the genome (background selection). The authors propose the latter – efficient purifying selection and strong linkage – as an explanation for the low genetic diversity observed in M. tuberculosis. This is of course not exclusive of other related explanations, such as clonal interference (Gerrish and Lenski 1998). They also champion the use of forward evolutionary simulations (Haller and Messer 2019) to model the interplay between selection, recombination, and demography as a powerful alternative to traditional backward coalescent models. At times, Stritt and Gagneux are pessimistic about our existing methods – arguing that dN/dS and homoplasies “tell us little about the frequency and strength of selection.” Even though I favour a more optimistic view, I fully agree that our traditional population genetic metrics are sensitive to a slew of different deviations from a standard neutral evolution model and must be interpreted with caution. As I and others have argued, the extent of recombination (measured as the amount of linkage in a genome) is a key factor in determining how best to test for natural selection (Shapiro et al. 2009) and to conduct genotype-phenotype association studies (Chen and Shapiro 2021) in microbes. While this article is focused on the well-studied M. tuberculosis complex, there are many parallels with other clonal bacteria, including pathogens and symbionts. Whatever your favourite bug, we must all be careful to make sure the stories we tell about them are not “just so tales” but are supported, to the extent possible, by data and quantitative models. References Chen, Peter E., and B. Jesse Shapiro. 2021. "Classic Genome-Wide Association Methods Are Unlikely to Identify Causal Variants in Strongly Clonal Microbial Populations." bioRxiv. Stritt, C., Gagneux, S. (2023). How do monomorphic bacteria evolve? The Mycobacterium tuberculosis complex and the awkward population genetics of extreme clonality. EcoEvoRxiv, ver.3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.32942/X2GW2P | How do monomorphic bacteria evolve? The *Mycobacterium tuberculosis* complex and the awkward population genetics of extreme clonality | Christoph Stritt, Sebastien Gagneux | <p style="text-align: justify;">Exchange of genetic material through sexual reproduction or horizontal gene transfer is ubiquitous in nature. Among the few outliers that rarely recombine and mainly evolve by <em>de novo</em> mutation are a group o... | | Evolutionary Dynamics, Genome Evolution, Molecular Evolution, Population Genetics / Genomics, Reproduction and Sex | B. Jesse Shapiro | Gonçalo Themudo | 2022-12-16 13:41:53 | |
30 May 2023
slendr: a framework for spatio-temporal population genomic simulations on geographic landscapesA new powerful tool to easily encode the geo-spatial dimension in population genetics simulationsRecommended by Emiliano Trucchi based on reviews by Liisa Loog and 2 anonymous reviewers
Models explaining the evolutionary processes operating in living beings are often impossible to test in the real world. This is mainly because of the long time (i.e., the number of generations) which is necessary for evolution to unfold. In addition, any such experiment would require a large number of individuals and, more importantly, many replicates to account for the inherent variance of the evolutionary processes under investigation. Only organisms with fast generation times and favourable rearing conditions can be used to explicitly test for specific evolutionary hypotheses. Computer simulations have filled this gap, revolutionising experimental testing in evolutionary biology by integrating genetic models into complex population dynamics, which can be run for (potentially) any length of time. Without going into an extensive description of the many available approaches for population genetics simulations (an exhaustive review can be found in Hoban et al 2012), three main aspects are, in my opinion, important for categorising and choosing one simulation approach over another. The first concerns the basic distinction between coalescent-based and individual-based simulators: the former being an efficient approach, which simulates back in time the coalescence events of a sample of homologous DNA fragments, while the latter is a more computationally intensive approach where all of the individuals (and their underlying genetic/genomic features) in the population are simulated forward-in-time, generation after generation. The second aspect concerns the simulation of natural selection. Although natural selection can be integrated into backward-in-time simulations, it is more realistically implemented as individual-based fitness in forward-in-time simulators. The third point, which has been often overlooked in evolutionary simulations, is about the possibility to design a simulation scenario where individuals and populations can exploit a physical (geographical) space. Amongst the coalescent-based simulators, SPLATCHE (Currat et al 2004), and its derivatives, is one of the few simulation tools deploying the coalescence process in sub-demes which are all connected by migration, thus getting as close as possible to a spatially-explicit population. On the other hand, individual-based simulators, whose development followed the increasing power of computational machines, offer a great opportunity to include spatio-temporal dynamics within a genomic simulation model. One of the most realistic and efficient individual-based forward-in-time simulators available is SLiM (Haller and Messer 2017), which allows users to implement simulations in arbitrarily complex spaces. Here, the more challenging part is encoding the spatially-explicit scenarios using the SLiM-specific EIDOS language. The new R package slendr (Petr et al 2022) offers a practical solution to this issue. By wrapping different tools into a well-known scripting language, slendr allows the design of spatiotemporal simulation scenarios which can be directly executed in the individual-based SLiM simulator, and the output stored with modern tree-sequence analysis tools (tskit; Kellerer et al 2018). Alternatively, simulations of non-spatial models can be run using a coalescent-based algorithm (msprime; Baumdicker et al 2022). The main advantage of slendr is that the whole simulative experiment can be performed entirely in the R environment, taking advantage of the many libraries available for geospatial and genomic data analysis, statistics, and visualisation. The open-source nature of this package, whose main aim is to make complex population genomics modelling more accessible, and the vibrant community of SLiM and tskit users will very likely make slendr widely used amongst the molecular ecology and evolutionary biology communities. Slendr handles real Earth cartographic data where users can design realistic demographic processes which characterise natural populations (i.e., expansions, displacement of large populations, interactions among populations, migrations, population splits, etc.) by changing spatial population boundaries across time and space. All in all, slendr is a very flexible and scalable framework to test the accuracy of spatial models, hypotheses about demography and selection, and interactions between organisms across space and time. REFERENCES Baumdicker, F., Bisschop, G., Goldstein, D., Gower, G., Ragsdale, A. P., Tsambos, G., ... & Kelleher, J. (2022). Efficient ancestry and mutation simulation with msprime 1.0. Genetics, 220(3), iyab229. https://doi.org/10.1093/genetics/iyab229 Currat, M., Ray, N., & Excoffier, L. (2004). SPLATCHE: a program to simulate genetic diversity taking into account environmental heterogeneity. Molecular Ecology Notes, 4(1), 139-142. https://doi.org/10.1046/j.1471-8286.2003.00582.x Haller, B. C., & Messer, P. W. (2017). SLiM 2: flexible, interactive forward genetic simulations. Molecular biology and evolution, 34(1), 230-240. https://doi.org/10.1093/molbev/msw211 Hoban, S., Bertorelle, G., & Gaggiotti, O. E. (2012). Computer simulations: tools for population and evolutionary genetics. Nature Reviews Genetics, 13(2), 110-122. https://doi.org/10.1038/nrg3130 Kelleher, J., Thornton, K. R., Ashander, J., & Ralph, P. L. (2018). Efficient pedigree recording for fast population genetics simulation. PLoS computational biology, 14(11), e1006581. https://doi.org/10.1371/journal.pcbi.1006581 Petr, M., Haller, B. C., Ralph, P. L., & Racimo, F. (2023). slendr: a framework for spatio-temporal population genomic simulations on geographic landscapes. bioRxiv, 2022.03.20.485041, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.03.20.485041 | slendr: a framework for spatio-temporal population genomic simulations on geographic landscapes | Martin Petr, Benjamin C. Haller, Peter L. Ralph, Fernando Racimo | <p style="text-align: justify;">One of the goals of population genetics is to understand how evolutionary forces shape patterns of genetic variation over time. However, because populations evolve across both time and space, most evolutionary proce... | | Bioinformatics & Computational Biology, Evolutionary Theory, Phylogeography & Biogeography, Population Genetics / Genomics | Emiliano Trucchi | 2022-09-14 12:57:56 | ||
22 May 2023
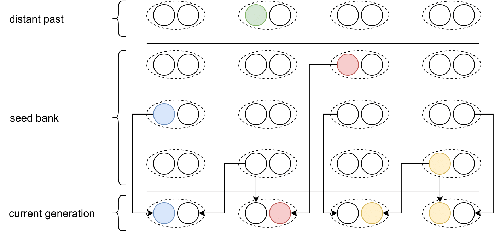
Weak seed banks influence the signature and detectability of selective sweepsNew insights into the dynamics of selective sweeps in seed-banked speciesRecommended by Renaud Vitalis based on reviews by Guillaume Achaz, Jere Koskela, William Shoemaker and Simon Boitard
Many organisms across the Tree of life have the ability to produce seeds, eggs, cysts, or spores, that can remain dormant for several generations before hatching. This widespread adaptive trait in bacteria, fungi, plants and animals, has a significant impact on the ecology, population dynamics and population genetics of species that express it (Evans and Dennehy 2005). In population genetics, and despite the recognition of its evolutionary importance in many empirical studies, few theoretical models have been developed to characterize the evolutionary consequences of this trait on the level and distribution of neutral genetic diversity (see, e.g., Kaj et al. 2001; Vitalis et al. 2004), and also on the dynamics of selected alleles (see, e.g., Živković and Tellier 2018). However, due to the complexity of the interactions between evolutionary forces in the presence of dormancy, the fate of selected mutations in their genomic environment is not yet fully understood, even from the most recently developed models. In a comprehensive article, Korfmann et al. (2023) aim to fill this gap by investigating the effect of germ banking on the probability of (and time to) fixation of beneficial mutations, as well as on the shape of the selective sweep in their vicinity. To this end, Korfmann et al. (2023) developed and released their own forward-in-time simulator of genome-wide data, including neutral and selected polymorphisms, that makes use of Kelleher et al.’s (2018) tree sequence toolkit to keep track of gene genealogies. The originality of Korfmann et al.’s (2023) study is to provide new quantitative results for the effect of dormancy on the time to fixation of positively selected mutations, the shape of the genomic landscape in the vicinity of these mutations, and the temporal dynamics of selective sweeps. Their major finding is the prediction that germ banking creates narrower signatures of sweeps around positively selected sites, which are detectable for increased periods of time (as compared to a standard Wright-Fisher population). The availability of Korfmann et al.’s (2023) code will allow a wider range of parameter values to be explored, to extend their results to the particularities of the biology of many species. However, as they chose to extend the haploid coalescent model of Kaj et al. (2001), further development is needed to confirm the robustness of their results with a more realistic diploid model of seed dormancy. REFERENCES Evans, M. E. K., and J. J. Dennehy (2005) Germ banking: bet-hedging and variable release from egg and seed dormancy. The Quarterly Review of Biology, 80(4): 431-451. https://doi.org/10.1086/498282 Kaj, I., S. Krone, and M. Lascoux (2001) Coalescent theory for seed bank models. Journal of Applied Probability, 38(2): 285-300. https://doi.org/10.1239/jap/996986745 Kelleher, J., K. R. Thornton, J. Ashander, and P. L. Ralph (2018) Efficient pedigree recording for fast population genetics simulation. PLoS Computational Biology, 14(11): e1006581. https://doi.org/10.1371/journal.pcbi.1006581 Korfmann, K., D. Abu Awad, and A. Tellier (2023) Weak seed banks influence the signature and detectability of selective sweeps. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.04.26.489499 Vitalis, R., S. Glémin, and I. Olivieri (2004) When genes go to sleep: the population genetic consequences of seed dormancy and monocarpic perenniality. American Naturalist, 163(2): 295-311. https://doi.org/10.1086/381041 Živković, D., and A. Tellier (2018). All but sleeping? Consequences of soil seed banks on neutral and selective diversity in plant species. Mathematical Modelling in Plant Biology, 195-212. https://doi.org/10.1007/978-3-319-99070-5_10 | Weak seed banks influence the signature and detectability of selective sweeps | Kevin Korfmann, Diala Abu Awad, Aurélien Tellier | <p style="text-align: justify;">Seed banking (or dormancy) is a widespread bet-hedging strategy, generating a form of population overlap, which decreases the magnitude of genetic drift. The methodological complexity of integrating this trait impli... | | Adaptation, Bioinformatics & Computational Biology, Evolutionary Applications, Evolutionary Ecology, Genome Evolution, Life History, Population Genetics / Genomics | Renaud Vitalis | 2022-05-23 13:01:57 | ||
16 May 2023
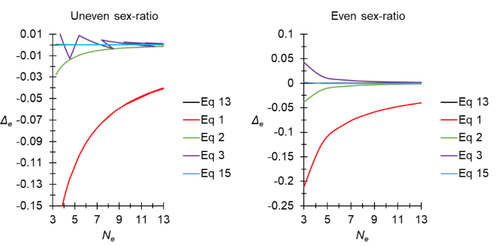
A new and almost perfectly accurate approximation of the eigenvalue effective population size of a dioecious population: comparisons with other estimates and detailed proofsAll you ever wanted to know about Ne in one handy placeRecommended by Charles Baer based on reviews by Jesse ("Jay") Taylor and 1 anonymous reviewerOf the four evolutionary forces, three can be straightforwardly summarized both conceptually and mathematically in the context of an allele at a genomic locus. Mutation (the mutation rate, μ) is simply captured by the per-site, per-generation probability that an allele mutates into a different allele. Recombination (the recombination rate, r) is captured as the probability of recombination between two sites, wherein alleles that are in different genomes in one generation come together in the same genome in the next generation. Natural selection (the selection coefficient, s) is captured by the probability that an allele is present in the next generation, relative to some reference. Random genetic drift – the random fluctuation in allele frequency due to sampling in a finite population - is not so straightforwardly summarized. The first, and most common way of characterizing evolutionary dynamics in a finite population is the Wright-Fisher model, in which the only deviation from the assumptions of Hardy-Weinberg conditions is finite population size. Importantly, in a W-F population, mating between diploid individuals is random, which implies self-fertile monoecy, and generations are non-overlapping. In an ideal W-F population, the probability that a gene copy leaves i descendants in the next generation is the result of binomial sampling of uniting gametes (if the locus is biallelic). The – and the next word is meaningful – magnitude/strength/rate/power/amount of genetic drift is proportional to 1/2N, where N is the size of the population. All of the following are affected by genetic drift: (1) the probability that a neutral allele ultimately reaches fixation, (2) the rate of loss of genetic variation within a population, (3) the rate of increase of genetic variance among populations, (4) the amount of genetic variation segregating in a population, (5) the probability of fixation/loss of a weakly selected variant. Presumably no real population adheres to ideal W-F conditions, which leads to the notion of "effective population size", Ne (Wright 1931), loosely defined as "the size of an ideal W-F population that experiences an equivalent strength of genetic drift". Almost always, Ne<N, and any violation of W-F assumptions can affect Ne. Importantly, Ne can be defined in different ways, and the specific formulation of Ne can have different implications for evolution. Ne was initially defined in terms of the rate of decrease of heterozygosity (inbreeding effective size) and increase in variance among populations (variance effective size). Ewens (1979) defined the Eigenvalue effective size (equivalent to the "random extinction" effective size) and elaborated on the conditions under which the various formulations of Ne differ (Ewens 1982). Nordborg and Krone (2002) defined the effective size in terms of the coalescent, and they identified conditions in which genetic drift cannot be described in terms of a W-F model (Sjodin et al. 2005); also see Karasov et al. (2010); Neher and Shraiman (2011). Distinct from the issue of defining Ne is the issue of calculating Ne from data, which is the focus of this paper by De Meeus and Noûs (2023). Pudovkin et al. (1996) showed that the Eigenvalue effective size in a dioecious population can be formulated in terms of excess heterozygosity, which the current authors note is equivalent to formulating Ne in terms of Wright's FIS statistic. As emphasized by the title, the marquee contribution of this paper is to provide a better approximation of the Eigenvalue effective size in a dioecious population. Science marches onward, although the empirical utility of this advance is obviously limited, given the tremendous inherent sources of uncertainty in real-world estimates of Ne. Perhaps more valuable, however, is the extensive set of appendixes, in which detailed derivations are provided for the various formulations of effective size. By way of analogy, the material presented here can be thought of as an extension of the material presented in section 7.6 of Crow and Kimura (1970), in which the Inbreeding and Variance effective population sizes are derived and compared. The appendixes should serve as a handy go-to source of detailed theoretical information with respect to the different formulations of effective population size. REFERENCES Crow, J. F. and M. Kimura. 1970. An Introduction to Population Genetics Theory. The Blackburn Press, Caldwell, NJ. De Meeûs, T. and Noûs, C. 2023. A new and almost perfectly accurate approximation of the eigenvalue effective population size of a dioecious population: comparisons with other estimates and detailed proofs. Zenodo, ver. 6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.5281/zenodo.7927968 Ewens, W. J. 1979. Mathematical Population Genetics. Springer-Verlag, Berlin. Ewens, W. J. 1982. On the concept of the effective population size. Theoretical Population Biology 21:373-378. https://doi.org/10.1016/0040-5809(82)90024-7 Karasov, T., P. W. Messer, and D. A. Petrov. 2010. Evidence that adaptation in Drosophila Is not limited by mutation at single sites. Plos Genetics 6. https://doi.org/10.1371/journal.pgen.1000924 Neher, R. A. and B. I. Shraiman. 2011. Genetic Draft and Quasi-Neutrality in Large Facultatively Sexual Populations. Genetics 188:975-U370. https://doi.org/10.1534/genetics.111.128876 Nordborg, M. and S. M. Krone. 2002. Separation of time scales and convergence to the coalescent in structured populations. Pp. 194–232 in M. Slatkin, and M. Veuille, eds. Modern Developments in Theoretical Population Genetics: The Legacy of Gustave Malécot. Oxford University Press, Oxford. https://www.webpages.uidaho.edu/~krone/malecot.pdf Pudovkin, A. I., D. V. Zaykin, and D. Hedgecock. 1996. On the potential for estimating the effective number of breeders from heterozygote-excess in progeny. Genetics 144:383-387. https://doi.org/10.1093/genetics/144.1.383 Sjodin, P., I. Kaj, S. Krone, M. Lascoux, and M. Nordborg. 2005. On the meaning and existence of an effective population size. Genetics 169:1061-1070. https://doi.org/10.1534/genetics.104.026799 Wright, S. 1931. Evolution in Mendelian populations. Genetics 16:0097-0159. https://doi.org/10.1093/genetics/16.2.97 | A new and almost perfectly accurate approximation of the eigenvalue effective population size of a dioecious population: comparisons with other estimates and detailed proofs | Thierry de Meeûs and Camille Noûs | <p>The effective population size is an important concept in population genetics. It corresponds to a measure of the speed at which genetic drift affects a given population. Moreover, this is most of the time the only kind of population size that e... | | Bioinformatics & Computational Biology, Evolutionary Ecology, Evolutionary Theory, Population Genetics / Genomics, Reproduction and Sex | Charles Baer | 2023-02-22 16:53:49 | ||
11 May 2023

Co-obligate symbioses have repeatedly evolved across aphids, but partner identity and nutritional contributions vary across lineagesFlexibility in Aphid Endosymbiosis: Dual Symbioses Have Evolved Anew at Least Six TimesRecommended by Olivier Tenaillon based on reviews by Alex C. C. Wilson and 1 anonymous reviewerIn this intriguing study (Manzano-Marín et al. 2022) by Alejandro Manzano-Marin and his colleagues, the association between aphids and their symbionts is investigated through meta-genomic analysis of new samples. These associations have been previously described as leading to fascinating genomic evolution in the symbiont (McCutcheon and Moran 2012). The bacterial genomes exhibit a significant reduction in size and the range of functions performed. They typically lose the ability to produce many metabolites or biobricks created by the host, and instead, streamline their metabolism by focusing on the amino acids that the host cannot produce. This level of co-evolution suggests a stable association between the two partners. However, the new data suggests a much more complex pattern as multiple independent acquisitions of co-symbionts are observed. Co-symbiont acquisition leads to a partition of the functions carried out on the bacterial side, with the new co-symbiont taking over some of the functions previously performed by Buchnera. In most cases, the new co-symbiont also brings the ability to produce B1 vitamin. Various facultative symbiotic taxa are recruited to be co-symbionts, with the frequency of acquisition related to the bacterial niche and lifestyle. REFERENCES Manzano-Marín, Alejandro, Armelle Coeur D’acier, Anne-Laure Clamens, Corinne Cruaud, Valérie Barbe, and Emmanuelle Jousselin. 2023. “Co-Obligate Symbioses Have Repeatedly Evolved across Aphids, but Partner Identity and Nutritional Contributions Vary across Lineages.” bioRxiv, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.08.28.505559. McCutcheon, John P., and Nancy A. Moran. 2012. “Extreme Genome Reduction in Symbiotic Bacteria.” Nature Reviews Microbiology 10 (1): 13–26. https://doi.org/10.1038/nrmicro2670. | Co-obligate symbioses have repeatedly evolved across aphids, but partner identity and nutritional contributions vary across lineages | Alejandro Manzano-Marín, Armelle Coeur d'acier, Anne-Laure Clamens, Corinne Cruaud, Valérie Barbe, Emmanuelle Jousselin | <p style="text-align: justify;">Aphids are a large family of phloem-sap feeders. They typically rely on a single bacterial endosymbiont, <em>Buchnera aphidicola</em>, to supply them with essential nutrients lacking in their diet. This association ... | | Genome Evolution, Other, Species interactions | Olivier Tenaillon | 2022-11-16 10:13:37 | ||
02 May 2023
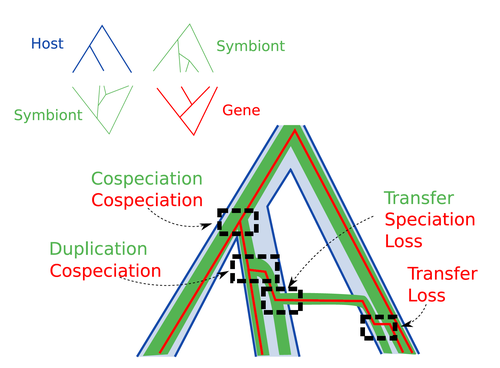
Host-symbiont-gene phylogenetic reconciliationReconciling molecular evolution and evolutionary ecology studies: a phylogenetic reconciliation method for gene-symbiont-host systemsRecommended by Emmanuelle Jousselin based on reviews by Vincent Berry and Catherine MatiasInteractions between species are a driving force in evolution. Many organisms host symbiotic partners that live all or part of their life in or on their host. Whether they are mutualistic or parasitic, these symbiotic associations impose strong selective pressures on both partners and affect their evolutionary trajectories. In fine, they can have a significant impact on the diversification patterns of both host and symbiont lineages, with symbiotic lineages sometimes speciating simultaneously with their hosts and/or switching from one host species to another. Long-term associations between species can also result in gene transfers between the involved organisms. Those lateral gene transfers are a source of ecological innovation but can obscure the phylogenetic signals and render the process of phylogenetic reconstructions complex (Lerat et al. 2003). Methods known as reconciliations explore similarities and differences between phylogenetic trees. They have been widely used to both compare the diversification patterns of hosts and symbionts and identify lateral gene transfers between species. Though the reconciliation approaches used in host/ symbiont and species/ gene phylogenetic studies are identical, they are always applied separately to solve either molecular evolution questions or investigate the evolution of ecological interactions. However, the two questions are often intimately linked and the current interest in multi-level systems (e.g. the holobiont concept) calls for a unique model that will take into account three-level nested organization (gene/symbiont/ host) where both symbiont and genes can transfer among hosts. Here Menet and collaborators (2023) provide such a model to produce three-level reconciliations. In order to do so, they extend the two-level reconciliation model implemented in “ALE” software (Szöllősi et al. 2013), one of the most used and proven reconciliation methods. Briefly, given a symbiont gene tree, a symbiont tree and a host tree, as in previous reconciliation models, the symbiont tree is mapped onto the host tree by mixing three types of events: Duplication, Transfer or Loss (DTL), with a possibility of the symbiont evolving temporarily outside the host phylogeny (in a “ghost” host lineage). The gene tree evolves similarly inside the symbiont tree, but horizontal transfers are constrained to symbionts co-occurring within the same host. Joint reconciliation scenarios are reconstructed and DTL event rates and likelihoods are estimated according to the model. As a nice addition, the authors propose a method to infer the symbiont phylogeny through amalgamation from gene trees and a host tree. The authors then explore the diverse possibilities offered by this method by testing it on both simulated datasets and biological datasets in order to check whether considering three nested levels is worthwhile. They convincingly show that three-level reconciliation has a better capacity to retrieve the symbiont donors and receivers of horizontal gene transfers, probably because transfers are constrained by additional elements relevant to the biological systems. Using, aphids, their obligate endosymbionts, and the symbiont genes involved in their nutritional functions, they identify horizontal gene transfers between aphid symbionts that are missed by two-level reconciliations but detected by expertise (Manzano-Marín et al. 2020). The other dataset presented here is on the human pathogen Helicobacter pylori, which history is supposed to reflect human migration. They use more than 1000 H. pylori gene families, and four populations, and use likelihood computations to compare different hypotheses on the diversification of the host. In summary, this study is a proof-of-concept of a 3-level reconciliation, where the authors manage to convey the applicability of their framework to many biological systems. Reported complexities, confirmed by reported running times, show that the method is computationally efficient. Without a doubt, the tool presented here will be very useful to evolutionary biologists who want to investigate multi-scale cophylogenies and it will move forward the study of associations between host and symbionts when symbiont genomic data are available. REFERENCES Lerat, E., Daubin, V., & Moran, N. A. (2003). From gene trees to organismal phylogeny in prokaryotes: the case of the γ-Proteobacteria. PLoS biology, 1(1), e19. Menet H, Trung AN, Daubin V, Tannier E (2023) Host-symbiont-gene phylogenetic reconciliation. bioRxiv, 2022.07.01.498457, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.07.01.498457 Szöllősi, G. J., Rosikiewicz, W., Boussau, B., Tannier, E., & Daubin, V. (2013). Efficient exploration of the space of reconciled gene trees. Systematic biology, 62(6), 901-912. | Host-symbiont-gene phylogenetic reconciliation | Hugo Menet, Alexia Nguyen Trung, Vincent Daubin, Eric Tannier | <p style="text-align: justify;"><strong>Motivation:</strong> Biological systems are made of entities organized at different scales e.g. macro-organisms, symbionts, genes...) which evolve in interaction.<br>These interactions range from indepe... | | Bioinformatics & Computational Biology, Phylogenetics / Phylogenomics | Emmanuelle Jousselin | 2022-08-21 18:34:27 | ||
02 May 2023
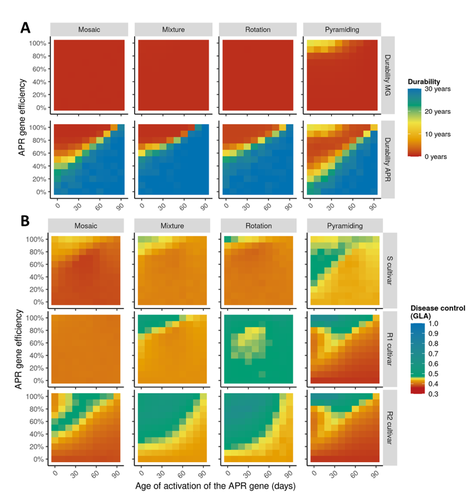
Durable resistance or efficient disease control? Adult Plant Resistance (APR) at the heart of the dilemmaPlant resistance to pathogens: just you wait?Recommended by Timothée Poisot based on reviews by Jean-Paul Soularue and 1 anonymous reviewerIn this preprint, Rimbaud et al. (2023) examine whether Adult Plant Resistance (APR), where plants delay their response to pathogens, is a viable alternative when the solution to evolve complete resistance from the seedling stage exists. At first glance, delaying resistance seems like a counter-intuitive strategy, unless it can result in a weaker selection of the pathogen, and therefore slow down its adaption to plant resistance. The approach of Rimbaud et al. is to incorporate as much of the mechanisms as possible into a model. By accounting for explicit spatio-temporal dynamics, stochasticity, and the coupling between demography and population genetics, to simulate an agricultural landscape, they reach a nuanced conclusion. Weaker and delayed activation of genes that confer APR does indeed reduce the selection pressure acting on the pathogen, at the cost of overall less effective protection. The alternative strategy of rapid or complete activation of these genes, although it results in better results in defending against the pathogen, is at risk of being overcome because it introduces a stronger selection pressure. One important feature of this work is that it accounts for agricultural practices. The landscape that is simulated can account for monoculture, mosaic cultures, mixed cultures, and rotations of crops (with different strategies for resistance). This introduces an interesting element to the conclusion: that human practices will have an impact on the selection pressures acting within the system. Perhaps the most striking result is that, for the plants, it might be more beneficial to bear the cost of a wild-type pathogen that can benefit from delayed activation of resistance, and therefore exclude the more virulent strains by simply being there first, and essentially buying the plant some time before it activates its resistance more completely. When the landscape is aggregated, even wild-type pathogens can cause severe epidemics; increasing fragmentation, because it enables connectivity between patches of plants with different strategies, allows pathogens to move across cultivars, and reduces the epidemic risk on susceptible plants. These results should encourage scaling up the perspective on APR, and indeed Rimbaud et al. adopt a landscape-scale perspective, to show that APR genes and genes conferring more complete resistance early on can have synergistic effects. This is, again, both an interesting result for evolutionary biologists, but also a useful way to prioritize different crop management strategies over large spatial scales. References Rimbaud, Loup, et al. Durable Resistance or Efficient Disease Control? Adult Plant Resistance (APR) at the Heart of the Dilemma. 2023. bioRxiv, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.08.30.505787 | Durable resistance or efficient disease control? Adult Plant Resistance (APR) at the heart of the dilemma | Loup Rimbaud, Julien Papaïx, Jean-François Rey, Benoît Moury, Luke G. Barrett, Peter H. Thrall | <p style="text-align: justify;">Adult plant resistance (APR) is an incomplete and delayed protection of plants against pathogens. At first glance, such resistance should be less efficient than classical major-effect resistance genes, which confer ... | | Adaptation, Evolutionary Applications, Evolutionary Epidemiology | Timothée Poisot | 2022-09-02 16:36:32 |




