
Latest recommendations
| Id | Title * | Authors * | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
02 Feb 2024
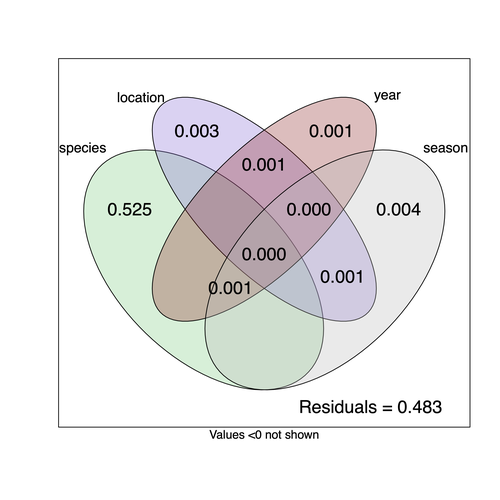
Community structure of heritable viruses in a Drosophila-parasitoids complexThe virome of a Drosophilidae-parasitoid communityRecommended by Ben Longdon based on reviews by 3 anonymous reviewers based on reviews by 3 anonymous reviewers
Understanding the factors that shape the virome of a host is key to understanding virus ecology and evolution (Obbard, 2018; French & Holmes, 2020). There is still much to learn about the diversity and distribution of viruses in a host community (Wille et al., 2019; Chen et al., 2023). The viruses of parasitoid wasps are well studied, and their viruses, or integrated viral genes, are known to suppress their insect host’s immune response to enhance parasitoid survival (Herniou et al., 2013; Coffman et al., 2022). Likewise, the insect virome is being increasingly well studied (Shi et al., 2016), with the virome of Drosophila species being particularly well characterised over the best part of the last century (L'Heritier & Teissier, 1937; L'Heritier, 1970; Brun & Plus, 1980; Longdon et al., 2010; Longdon et al., 2011; Longdon et al., 2012; Webster et al., 2015; Webster et al., 2016; Medd et al., 2018; Wallace et al., 2021). However, the viromes of parasitoids and their insect host communities have been less well studied (Leigh et al., 2018; Caldas-Garcia et al., 2023), and the inherent connectivity between parasitoids and their hosts provides an interesting system to study virus host range and cross-species transmission. Here, Varaldi et al (Varaldi et al., 2024) have examined the viruses associated with a community of nine Drosophilidae hosts and six parasitoids. Using both RNA and DNA sequencing of insects reared for two generations, they selected viruses that are maintained in the lab either via vertical transmission or contamination of rearing medium. From 55 pools of insects they found 53 virus-like sequences, 37 of which were novel. Parasitoids were host to nearly twice as many viruses as their Drosophila hosts, although they note this could be due to differences in the rearing temperatures of the hosts. They next quantified if species, year, season, or location played a role in structuring the virome, finding only a significant effect of host species, which explained just over 50% of the variation in virus distribution. No evidence was found of related species sharing more similar virus communities. Although looking at a limited number of species, this suggests that these viruses are not co-speciating or preferentially host switching between closely related species. Finally, they carried out crosses between lines of the parasitoid Leptopilina heterotoma that were infected and uninfected for a novel Iflavirus found in their sequencing data. They found evidence of high levels of maternal transmission and lower level horizontal transmission between wasp larvae parasitising the same host. No evidence of changes in parasitoid-induced mortality, developmental success or the sex ratio was found in iflavirus-infected parasitoids. Interestingly individuals infected with this RNA virus also contained viral DNA, but this did not appear to be integrated into the wasp genome. Overall, this work has taken the first steps in examining the community structure of the virome of parasitoids together with their Drosophilidae hosts. This work will not doubt stimulate follow-up studies to explore the evolution and ecology of these novel virus communities. References Brun G, Plus N (1980) The viruses of Drosophila. In: The genetics and biology of Drosophila eds Ashburner M & Wright TRF), pp. 625-702. Academic Press, New York. | Community structure of heritable viruses in a *Drosophila*-parasitoids complex | Julien Varaldi, David Lepetit, Nelly Burlet, Camille Faber, Bérénice Baretje, Roland Allemand | <p style="text-align: justify;">The diversity and phenotypic impacts related to the presence of heritable bacteria in insects have been extensively studied in the last decades. On the contrary, heritable viruses have been overlooked for several re... | | Evolutionary Ecology, Species interactions | Ben Longdon | 2023-08-03 01:07:43 | ||
25 Jan 2024

Sperm production and allocation in response to risk of sperm competition in the black soldier fly Hermetia illucensElevated sperm production and faster transfer: plastic responses to the risk of sperm competition in males of the black sodier fly Hermetia illuceRecommended by Trine Bilde based on reviews by Rebecca Boulton, Isabel Smallegange and 1 anonymous reviewer
In this paper (Manas et al., 2023), the authors investigate male responses to risk of sperm competition in the black soldier fly Hermetia illuce, a widespread insect that has gained recent attention for its potential to be farmed for sustainable food production (Tomberlin & van Huis, 2020). Using an experimental approach that simulated low-risk (males were kept individually) and high-risk (males were kept in groups of 10) of sperm competition, they found that males reared in groups showed a significant increase in sperm production compared with males reared individually. This shows a response to the rearing environment in sperm production that is consistent with an increase in the perceived risk of sperm competition. These males were then used in mating experiments to determine whether sperm allocation to females during mating was influenced by the perceived risk of sperm competition. Mating experiments were initiated in groups, since mating only occurs when more than one male and one female are present, indicating strong sexual selection in the wild. Once a copulation began, the pair was moved to a new environment with no competition, with male competitors, or with other females, to test how social environment and potentially the sex of surrounding individuals influenced sperm allocation during mating. Copulation duration and the number of sperm transferred were subsequently counted. In these mating experiments, the number of sperm stored in the female spermathecae increased under immediate risk of sperm competition. Interestingly, this was not because males copulated for longer depending on the risk of sperm competition, indicating that males respond plastically to the risk of competition by elevating their investment in sperm production and speed of sperm transfer. There was no difference between competitive environments consisting of males or females respectively, suggesting that it is the presence of other flies per se that influence sperm allocation. The study provides an interesting new example of how males alter reproductive investment in response to social context and sexual competition in their environment. In addition, it provides new insights into the reproductive biology of the black soldier fly Hermetia illucens, which may be relevant for optimizing farming conditions. References Manas F, Labrousse C, Bressac C (2023) Sperm production and allocation in response to risks of sperm competition in the black soldier fly Hermetia illucens. bioRxiv, 2023.06.20.544772, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.06.20.544772 Tomberlin JK, Van Huis A (2020) Black soldier fly from pest to ‘crown jewel’ of the insects as feed industry: an historical perspective. Journal of Insects as Food and Feed, 6, 1–4. https://doi.org/10.3920/JIFF2020.0003 | Sperm production and allocation in response to risk of sperm competition in the black soldier fly Hermetia illucens | Frédéric Manas, Carole Labrousse, Christophe Bressac | <p style="text-align: justify;">In polyandrous species, competition between males for offspring paternity goes on after copulation through the competition of their ejaculates for the fertilisation of female's oocytes. Given that males allocating m... | | Reproduction and Sex, Sexual Selection | Trine Bilde | 2023-06-26 09:41:07 | ||
08 Jan 2024
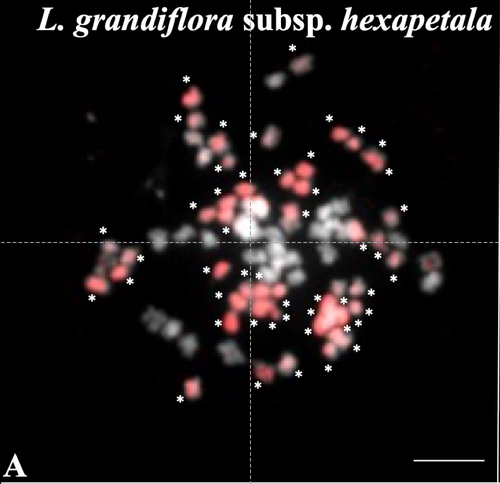
Genomic relationships among diploid and polyploid species of the genus Ludwigia L. section Jussiaea using a combination of molecular cytogenetic, morphological, and crossing investigationsDeciphering the genomic composition of tetraploid, hexaploid and decaploid Ludwigia L. species (section Jussiaea)Recommended by Malika AINOUCHE based on reviews by Alex BAUMEL and Karol MARHOLDPolyploidy, which results in the presence of more than two sets of homologous chromosomes represents a major feature of plant genomes that have undergone successive rounds of duplication followed by more or less rapid diploidization during their evolutionary history. Polyploid complexes containing diploid and derived polyploid taxa are excellent model systems for understanding the short-term consequences of whole genome duplication, and have been particularly well-explored in evolutionary ecology (Ramsey and Ramsey 2014, Rice et al. 2019). Many polyploids (especially when resulting from interspecific hybridization, i.e. allopolyploids) are successful invaders (te Beest et al. 2012) as a result of rapid genome dynamics, functional novelty, and trait evolution. The origin (parental legacy) and modes of formation of polyploids have a critical impact on the subsequent polyploid evolution. Thus, elucidation of the genomic composition of polyploids is fundamental to understanding trait evolution, and such knowledge is still lacking for many invasive species. Genus Ludwigia is characterized by a complex taxonomy, with an underexplored evolutionary history. Species from section Jussieae form a polyploid complex with diploids, tetraploids, hexaploids, and decaploids that are notorious invaders in freshwater and riparian ecosystems (Thouvenot et al.2013). Molecular phylogeny of the genus based on nuclear and chloroplast sequences (Liu et al. 2027) suggested some relationships between diploid and polyploid species, without fully resolving the question of the parentage of the polyploids. In their study, Barloy et al. (2023) have used a combination of molecular cytogenetics (Genomic In situ Hybridization), morphology and experimental crosses to elucidate the genomic compositions of the polyploid species, and show that the examined polyploids are of hybrid origin (allopolyploids). The tetraploid L. stolonifera derives from the diploids L. peploides subsp. montevidensis (AA genome) and L. helminthorhiza (BB genome). The tetraploid L. ascendens also share the BB genome combined with an undetermined different genome. The hexaploid L. grandiflora subsp. grandiflora has inherited the diploid AA genome combined with additional unidentified genomes. The decaploid L. grandiflora subsp. hexapetala has inherited the tetraploid L. stolonifera and the hexaploid L. grandiflora subsp. hexapetala genomes. As the authors point out, further work is needed, including additional related diploid (e.g. other subspecies of L. peploides) or tetraploid (L. hookeri and L. peduncularis) taxa that remain to be investigated, to address the nature of the undetermined parental genomes mentioned above. The presented work (Barloy et al. 2023) provides significant knowledge of this poorly investigated group with regard to genomic information and polyploid origin, and opens perspectives for future studies. The authors also detect additional diagnostic morphological traits of interest for in-situ discrimination of the taxa when monitoring invasive populations. References Barloy D., Portillo-Lemus L., Krueger-Hadfield S.A., Huteau V., Coriton O. (2024). Genomic relationships among diploid and polyploid species of the genus Ludwigia L. section Jussiaea using a combination of molecular cytogenetic, morphological, and crossing investigations. BioRxiv, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology https://doi.org/10.1101/2023.01.02.522458 te Beest M., Le Roux J.J., Richardson D.M., Brysting A.K., Suda J., Kubešová M., Pyšek P. (2012). The more the better? The role of polyploidy in facilitating plant invasions. Annals of Botany, Volume 109, Issue 1 Pages 19–45, https://doi.org/10.1093/aob/mcr277 Ramsey J. and Ramsey T. S. (2014). Ecological studies of polyploidy in the 100 years following its discovery Phil. Trans. R. Soc. B369 1–20 https://doi.org/10.1098/rstb.2013.0352 Rice, A., Šmarda, P., Novosolov, M. et al. (2019). The global biogeography of polyploid plants. Nat Ecol Evol 3, 265–273. https://doi.org/10.1038/s41559-018-0787-9 Thouvenot L, Haury J, Thiebaut G. (2013). A success story: Water primroses, aquatic plant pests. Aquat. Conserv. Mar. Freshw. Ecosyst. 23:790–803 https://doi.org/10.1002/aqc.2387 | Genomic relationships among diploid and polyploid species of the genus *Ludwigia* L. section *Jussiaea* using a combination of molecular cytogenetic, morphological, and crossing investigations | D. Barloy, L. Portillo - Lemus, S. A. Krueger-Hadfield, V. Huteau, O. Coriton | <p>ABSTRACTThe genus Ludwigia L. sectionJussiaeais composed of a polyploid species complex with 2x, 4x, 6x and 10x ploidy levels, suggesting possible hybrid origins. The aim of the present study is to understand the genomic relationships among dip... | | Hybridization / Introgression, Phylogenetics / Phylogenomics | Malika AINOUCHE | 2023-01-11 13:47:18 | ||
14 Dec 2023

Genetic sex determination in three closely related hydrothermal vent gastropods, including one species with intersex individualsA shared XY sex chromosome system with variable recombination ratesRecommended by Tanja Schwander based on reviews by Hugo Darras, Daniel Jeffries and 1 anonymous reviewerMany species with separate sexes have evolved sex chromosomes, with the sex-limited chromosomes (i.e. the Y or W chromosomes) exhibiting a wide range of genetic divergences from their homologous X or Z chromosomes (Bachtrog et al., 2014). Variable divergences can result from the cessation of recombination between sex chromosomes that occurred at different time points, with the mechanisms of initiation and expansion of recombination suppression along sex chromosomes remaining poorly understood (Charlesworth, 2017). The study by Castel et al (2023) describes the serendipitous discovery of a shared XY sex chromosome system in three closely related hydrothermal vent gastropods. The X and Y chromosomes appear to still recombine but at variable rates across the three species. This variation makes the gastropod system a very promising focus for future research on sex chromosome evolution. An additional intriguing finding is that some females in one of three gastropod species contain male reproductive tissue in their gonads, providing a fascinating case of a mixed or transitory sexual system. Overall, the study by Castel et al (2023) offers the first insights into the reproduction and sex chromosome system of animals living in deep marine vents, which have remained poorly studied and open outstanding research perspectives on these creatures. References Bachtrog, D., J.E.Mank, C.L.Peichel, M.Kirkpatrick, S.P.Otto, T.L. Ashman, M.W.Hahn, J.Kitano, I.Mayrose, R.Ming, et al. 2014.Sex determination: why so many ways of doing it? PLoSBiol. 12:e1001899. https://doi.org/10.1371/journal.pbio.1001899 Charlesworth, D. Young sex chromosomes in plants and animals. 2019. New Phytologist 224: 1095–1107. https://doi.org/10.1111/nph.16002 Castel J, Pradillon F, Cueff V, Leger G, Daguin-Thiébaut C, Ruault S, Mary J, Hourdez S, Jollivet D, and Broquet T 2023. Genetic sex determination in three closely related hydrothermal vent gastropods, including one species with intersex individuals. bioRxiv, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.04.11.536409 | Genetic sex determination in three closely related hydrothermal vent gastropods, including one species with intersex individuals | Castel J, Pradillon F, Cueff V, Leger G, Daguin-Thiébaut C, Ruault S, Mary J, Hourdez S, Jollivet D, and Broquet T | <p style="text-align: justify;">Molluscs have a wide variety of sexual systems and have undergone many transitions from separate sexes to hermaphroditism or vice versa, which is of interest for studying the evolution of sex determination and diffe... | | Population Genetics / Genomics, Reproduction and Sex | Tanja Schwander | 2023-04-14 11:48:25 | ||
29 Nov 2023
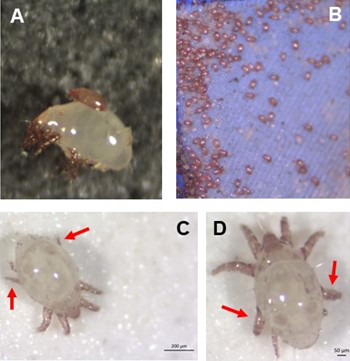
Individual differences in developmental trajectory leave a male polyphenic signature in bulb mite populationsWhat determines whether to scramble or fight in male bulb mitesRecommended by Ulrich Karl Steiner based on reviews by 2 anonymous reviewers
A classic textbook example in evolutionary ecology for phenotypic plasticity—the expression of different phenotypes by a genotype under different environmental conditions—is on Daphnia (Dodson 1989). If various species of this small crustacean are exposed to predation risk or cues thereof, their offspring show induced defense phenotypes including helmets, neck teeth, or head- and tail-spines. These induced morphological changes lower the risk of being eaten by a predator. As in Daphnia, induction can span over generations, while other induced phenotypic plastic changes are almost instantaneous, including many responses in physiology and behaviour (Gabriel et al. 2005). Larvae male bulb mites also show plasticity in morphologies throughout development. They can develop into a costly adult fighter morphology or a less costly, but vulnerable, scrambler type. The question Deere and Smallegange (Deere & Smallegange 2023) address is whether male bulb mite larvae can anticipate which type will likely be adaptive once they become adult, or alternatively, whether the resource availability or population density they experience during their larvae phase determines frequencies of adult scrambler and fighter types. They explore this question through experimental evolution, by removing different fractions of developing intermediate larva types. They thereby manipulate the stage structure of populations and alter selective forces on these stages. The potential shift of fixed genetics, imposed by the experimental selection regimes, is evaluated by fitness assays in green garden experiments. The exciting extension to classical experiments on phenotypic plasticity is that the authors aim at exploring eco-evolutionary feedbacks experimentally in a system that is a little more complex than basic host-parasite or predator-prey systems. The latter involve, for instance, rotifer-algae dynamics (Yoshida et al. 2003; Becks et al. 2012) or similar simple lab systems for which eco-evolutionary feedbacks have been demonstrated. The challenge for the exploration of more complex systems is revealed in the study by Deere and Smallegange. Their findings suggest that the frequencies of adult male morphotype is triggered by the environmental condition (nutrient availability) during the larval phase, i.e. a simple environmental induced plastic response. No fixed genetic shift in determining adult morphotype frequencies occurs. The trigger at the larva phase remains also not perfectly determined in their experiments, as population density and resource (food) availability are partly confounded. Additional complexity and selective aspects come into play in this system, as the targeted developmental stages that develop into fighter male morphs are also dispersal morphs. If selection on dispersal to avoid residing in food-limited environments is strong, triggering genetic shifts in fighter morphs by local population structure might be hard to experimentally achieve. Small sample sizes limit conclusions on complex interactions among duration of the experiment, population size, developmental stage types, and adult fighter frequencies. The presented study (Deere & Smallegange 2023) helps to bridge theoretical predictions and empirical evidence for eco-evolutionary feedbacks that goes beyond simple ecological-driven changes in population dynamics (Govaert et al. 2019).
References
Becks, L., Ellner, S.P., Jones, L.E. & Hairston, N.G. (2012). The functional genomics of an eco-evolutionary feedback loop: linking gene expression, trait evolution, and community dynamics. Ecol. Lett., 15, 492-501. | Individual differences in developmental trajectory leave a male polyphenic signature in bulb mite populations | Jacques A. Deere & Isabel M. Smallegange | <p style="text-align: justify;">Developmental plasticity alters phenotypes and can in that way change the response to selection. When alternative phenotypes show different life history trajectories, developmental plasticity can also affect, and be... | | Evolutionary Ecology, Life History, Phenotypic Plasticity, Sexual Selection | Ulrich Karl Steiner | 2023-02-07 12:14:33 | ||
29 Nov 2023

Does sociality affect evolutionary speed?On the evolutionary implications of being a social animalRecommended by Michael D Greenfield based on reviews by Rafael Lucas Rodriguez and 1 anonymous reviewerWhat does it mean to be highly social? Considering the so-called four ‘pinnacles’ of animal society (Wilson, 1975) – humans, cooperative breeding as found in some non-human mammals and birds, the social insects, and colonial marine invertebrates – having inter-individual relations extending beyond the sexual pair and the parent-offspring interaction is foremost. In many cases being social implies a high local population density, interaction with the same group of individuals over an extended time period, and an overlapping of generations. Additional features of social species may be a wide geographical range, perhaps associated with ecological and behavioral plasticity, the latter often facilitated by cultural transmission of traditions. Narrowing our perspective to the domain of PCI Evolutionary Biology, we might continue our question by asking whether being social predisposes one to a special evolutionary path toward the future. Do social species evolve faster (or slower) than their more solitary relatives such that over time they are more unlike (or similar to) those relatives (anagenesis)? And are evolutionary changes in social species more or less likely to be accompanied by lineage splitting (cladogenesis) and ultimately speciation? The latter question is parallel to one first posed over 40 years ago (West-Eberhard, 1979; Lande, 1981) for sexually selected traits: Do strong mating preferences and conspicuous courtship signals generate speciation via the Fisherian process or ecological divergence? An extensive survey of birds had found little supporting evidence (Price, 1998), but a recent one that focused on plumage complexity in tanagers did reveal a relationship, albeit a weak one (Price-Waldman et al., 2020). Because sexual selection has been viewed as a part of the broader process of social selection (West-Eberhard, 1979), it is thus fitting to extend our surveys to the evolutionary implications of being social. Unlike the inquiry for a sexual selection - evolutionary change connection, a social behavior counterpart has remained relatively untreated. Diverse logistical problems might account for this oversight. What objective proxies can be used for social behavior, and for the rate of evolutionary change within a lineage? How many empirical studies have generated data from which appropriate proxies could be extracted? More intractable is the conundrum arising from the connectedness between socially- and sexually-selected traits. For example, the elevated population density found in highly social species can greatly increase the mating advantage enjoyed by an attractive male. If anagenesis is detected, did it result from social behavior or sexual selection? And if social behavior leads to a group structure in which male-male competition is reduced, would a modest rate of evolutionary change be support for the sexual selection - evolutionary speed connection or evidence opposing the sociality - evolution one? Against the above odds, several biologists have begun to explore the notion that social behavior just might favor evolutionary speed in either anagenesis or cladogenesis. In a recent analysis relying on the comparative method, Lluís Socias-Martínez and Louise Rachel Peckre (2023) combed the scientific literature archives and identified those studies with specific data on the relationships between sexual selection or social behavior and evolutionary change, either anagenesis or cladogenesis. The authors were careful to employ fairly conservative criteria for including studies, and the number eventually retained was small. Nonetheless, some patterns emerge: Many more studies report anagenesis than cladogenesis, and many more report correlations with sexually-selected traits than with non-sexual social behavior ones. And, no study indicates a potential effect of social behavior on cladogenesis. Is this latter observation authentic or an artifact of a paucity of data? There are some a priori reasons why cladogenesis may seldom arise. Whereas highly social behavior could lead to fission encompassing mutually isolated population clusters within a species, social behavior may also engender counterbalancing plasticity that allows and even promotes inter-cluster migration and fusion. And briefly – and non-systematically, as the rate of lineage splitting would need to be measured – looking at one of the pinnacles of animal social behavior, the social insects, there is little indication that diversification has been accelerated. There are fewer than 3000 described species of termites, only ca. 16,000 ants, and the vast majority of bees and wasps are solitary. Lluís Socias-Martínez and Louise Rachel Peckre provide us with a very detailed discussion of these and a myriad of other complications. I end with a common refrain, we need more consideration of the authors’ interesting question, and much more data and analysis. One can thank Socias-Martínez and Peckre for pointing us in that direction. References Lande, R. (1981). Models of speciation by sexual selection on polygenic traits. Proc. Natn. Acad. Sci. USA 78, 3721-3725. https://doi.org/10.1073/pnas.78.6.3721 Price, T. (1998). Sexual selection and natural selection in bird speciation. Phil. Trans. Roy. Soc. B, 353, 251-260. https://doi.org/10.1098/rstb.1998.0207 Price‐Waldman, R. M., Shultz, A. J., & Burns, K. J. (2020). Speciation rates are correlated with changes in plumage color complexity in the largest family of songbirds. Evolution, 74(6), 1155–1169. https://doi.org/10.1111/evo.13982 Socias-Martínez and Peckre. (2023). Does sociality affect evolutionary speed? Zenodo, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.5281/zenodo.10086186 West-Eberhard, M. J. (1979). Sexual selection, social competition, and evolution. Proceedings of the American Philosophical Society, 123(4), 222–234. http://www.jstor.org/stable/2828804 Wilson, E. O. (1975). Sociobiology. The New Synthesis. Cambridge, Mass., The Belknap Press of Harvard University | Does sociality affect evolutionary speed? | Lluís Socias-Martínez, Louise Rachel Peckre | <p>An overlooked source of variation in evolvability resides in the social lives of animals. In trying to foster research in this direction, we offer a critical review of previous work on the link between evolutionary speed and sociality. A first ... | | Behavior & Social Evolution, Evolutionary Dynamics, Evolutionary Theory, Genome Evolution, Macroevolution, Molecular Evolution, Population Genetics / Genomics, Sexual Selection, Speciation | Michael D Greenfield | 2023-03-03 00:10:49 | ||
20 Nov 2023
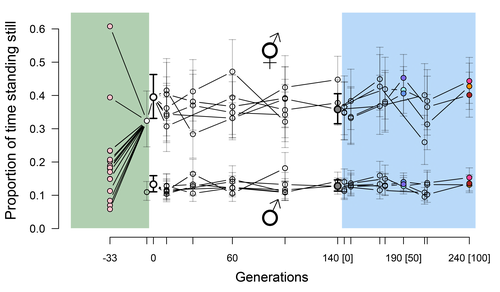
Phenotypic stasis with genetic divergencePhenotypic stasis despite genetic divergence and differentiation in Caenorhabditis elegans.Recommended by Frédéric Guillaume based on reviews by Benoit Pujol and Pedro Simões
Explaining long periods of evolutionary stasis, the absence of change in trait means over geological times, despite the existence of abundant genetic variation in most traits has challenged evolutionary theory since Darwin's theory of evolution by gradual modification (Estes & Arnold 2007). Stasis observed in contemporary populations is even more daunting since ample genetic variation is usually coupled with the detection of selection differentials (Kruuk et al. 2002, Morrissey et al. 2010). Moreover, rapid adaptation to environmental changes in contemporary populations, fuelled by standing genetic variation provides evidence that populations can quickly respond to an adaptive challenge. Explanations for evolutionary stasis usually invoke stabilizing selection as a main actor, whereby optimal trait values remain roughly constant over long periods of time despite small-scale environmental fluctuations. Genetic correlation among traits may also play a significant role in constraining evolutionary changes over long timescales (Schluter 1996). Yet, genetic constraints are rarely so strong as to completely annihilate genetic changes, and they may evolve. Patterns of genetic correlations among traits, as captured in estimates of the G-matrix of additive genetic co-variation, are subject to changes over generations under the action of drift, migration, or selection, among other causes (Arnold et al. 2008). Therefore, under the assumption of stabilizing selection on a set of traits, phenotypic stasis and genetic divergence in patterns of trait correlations may both be observed when selection on trait correlations is weak relative to its effect on trait means. Mallard et al. (2023) set out to test whether selection or drift may explain the divergence in genetic correlation among traits in experimental lines of the nematode Caenorhabditis elegans and whether stabilizing selection may be a driver of phenotypic stasis. To do so, they analyzed the evolution of locomotion behavior traits over 100 generations of lab evolution in a constant and homogeneous environment after 140 generations of domestication from a largely differentiated set of founder populations. The locomotion traits were transition rates between movement states and direction (still, forward or backward movement). They could estimate the traits' broad-sense G-matrix in three populations at two generations (50 and 100), and in the ancestral mixed population. Similarly, they estimated the shape of the selection surface by regressing locomotion behavior on fertility. Armed with both G-matrix and surface estimates, they could test whether the G's orientation matched selection's orientation and whether changes in G were constrained by selection. They found stasis in trait mean over 100 generations but divergence in the amount and orientation of the genetic variation of the traits relative to the ancestral population. The selected populations changed orientation of their G-matrices and lost genetic variation during the experiment in agreement with a model of genetic drift on quantitative traits. Their estimates of selection also point to mostly stabilizing selection on trait combinations with weak evidence of disruptive selection, suggesting a saddle-shaped selection surface. The evolutionary responses of the experimental populations were mostly consistent with small differentiation in the shape of G-matrices during the 100 generations of stabilizing selection. Mallard et al. (2023) conclude that phenotypic stasis was maintained by stabilizing selection and drift in their experiment. They argue that their findings are consistent with a "table-top mountain" model of stabilizing selection, whereby the population is allowed some wiggle room around the trait optimum, leaving space for random fluctuations of trait variation, and especially trait co-variation. The model is an interesting solution that might explain how stasis can be maintained over contemporary times while allowing for random differentiation of trait genetic co-variation. Whether such differentiation can then lead to future evolutionary divergence once replicated populations adapt to a new environment is an interesting idea to follow. References Arnold, S. J., Bürger, R., Hohenlohe, P. A., Ajie, B. C. and Jones, A. G. 2008. Understanding the evolution and stability of the G-matrix. Evolution 62(10): 2451-2461. | Phenotypic stasis with genetic divergence | François Mallard, Luke Noble, Thiago Guzella, Bruno Afonso, Charles F. Baer, Henrique Teotónio | <p style="text-align: justify;">Whether or not genetic divergence in the short-term of tens to hundreds of generations is compatible with phenotypic stasis remains a relatively unexplored problem. We evolved predominantly outcrossing, genetically ... | | Adaptation, Behavior & Social Evolution, Experimental Evolution, Quantitative Genetics | Frédéric Guillaume | 2022-09-01 14:32:53 | ||
13 Nov 2023
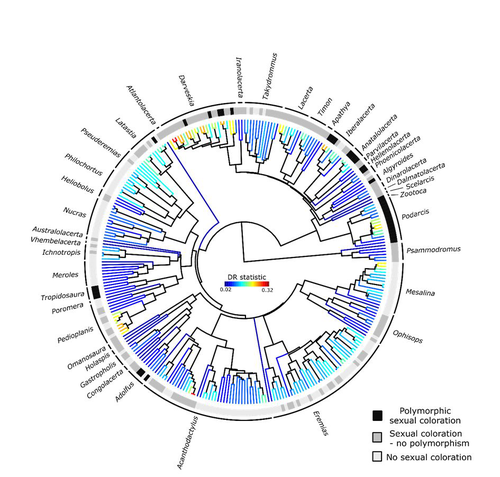
Color polymorphism and conspicuousness do not increase speciation rates in LacertidsColour polymorphism does not increase diversification rates in lizardsRecommended by Alejandro Gonzalez-Voyer based on reviews by 2 anonymous reviewers
The striking differences in species richness among lineages in the Tree of Life have long attracted much research interest. In particular, researchers have asked whether certain traits are associated with greater diversification, with a particular focus on traits under sexual selection given their direct link to mating isolation. Polymorphism, defined as the presence of co-occurring, heritable morphs within a population, has been proposed to influence diversification rates although the effect has been proposed as both promoting or alternatively impeding speciation. The effect of polymorphism may be positive, that is facilitating speciation if polymorphism allows to broaden the ecological niche, thus enabling range expansion, or enabling maintenance of populations in variable environments. Specialized ectomorphs have been observed in several species (e.g. Kusche et al. 2015, Lattanzio and Miles 2016, Whitney et al. 2018, Scali et al. 2016). Polymorphism may also facilitate speciation if a morph is lost during the colonization of a novel area or niche, resulting in rapid divergence of the remaining morphs and reproductive isolation from the ancestral population, known as the morph speciation hypothesis (West-Eberhard 1986, Corl et al. 2010). On the other hand, polymorphism may hamper speciation through disassortative maintaining by morph, which may maintain the polymorphism through the speciation process (Jamie and Meier 2020). An example of such a process is Heliconius numata where disassortative mate preferences based on color hampers ecological speciation (Chouteau et al. 2017). Previous evidence in birds and lizards suggests polymorphism favors diversification (Corl et al. 2010b, 2012, Hugall and Stuart-Fox 2012, Brock et al. 2021). Here, de Solan et al. (2023) test the effect of polymorphism on diversification in Lacertidae, a family of lizards containing more than 300 species distributed across Europe, Africa and Asia. The group offers a good model system to test the effect of polymorphism on speciation as it contains several species with colour polymorphism, sometimes present in both sexes but restricted to males when present in the flank. Using coloration data from the literature as well as photographs of live specimens for 295 species the authors tested whether the presence of polymorphism is associated with higher diversification rates. While undertaking their project, another group independently tackled the same question (Brock et al. 2021), using the same model system but coming to very different conclusions. Therefore, de Solan et al. (2023) decided to also contrast their results with those of Brock et al. (2021) to determine the factors responsible for the contrasting results of both studies. The latter I consider one of the strengths of the work, given the careful re-analyses to determine the causes of the discrepancies between both studies. De Solan et al. (2023) found no association between the presence of polymorphism and diversification rates, even though they used different analytical approaches. Thus, this study is interesting as it provides results that do not support a positive effect of polymorphism on species richness. The use of a phylogeny with more limited species sampling (García-Porta et al. 2019) implied that the authors had to manually add 75 species, of which 17 were added to the tree based on information from previously published trees and 68 were added at random locations within the genus. To control for potential biases the authors repeated the analyses using a sample of trees with the imputed taxa, results were broadly concordant across the set of trees. The careful re-analysis contrasting Brock et al. (2021) and de Solan et al. (2023) results suggests the difference is mainly due to a difference in how species were coded as presenting polymorphism, which differed between the two studies, as well as a difference in the package version used to run the state-dependent diversification models. Interestingly non-parametric analyses yielded similar results across both datasets. Garcia-Porta, J., Irisarri, I., Kirchner, M. et al. 2019. Environmental temperatures shape thermal physiology as well as diversification and genome-wide substitution rates in lizards. Nature Communications. 10: 4077. https://doi.org/10.1038/s41467-019-11943-x de Solan T, Sinervo B, Geniez P, David P, Crochet P-A (2023) Colour polymorphism and conspicuousness do not increase speciation rates in Lacertids. bioRxiv, 2023.02.15.528678, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.02.15.528678 West-Eberhard, M.J. 1986. Alternative adaptations, speciation, and phylogeny (A review). Proceedings of the National Academy of Sciences. 83: 1388-1392. https://doi.org/10.1073/pnas.83.5.1388 | Color polymorphism and conspicuousness do not increase speciation rates in Lacertids | Thomas de Solan, Barry Sinervo, Philippe Geniez, Patrice David, Pierre-André Crochet | <p style="text-align: justify;">Conspicuous body colors and color polymorphism have been hypothesized to increase rates of speciation. Conspicuous colors are evolutionary labile, and often involved in intraspecific sexual signaling and thus may pr... | | Evolutionary Ecology, Macroevolution, Speciation | Alejandro Gonzalez-Voyer | 2023-02-22 10:05:03 | ||
30 Oct 2023
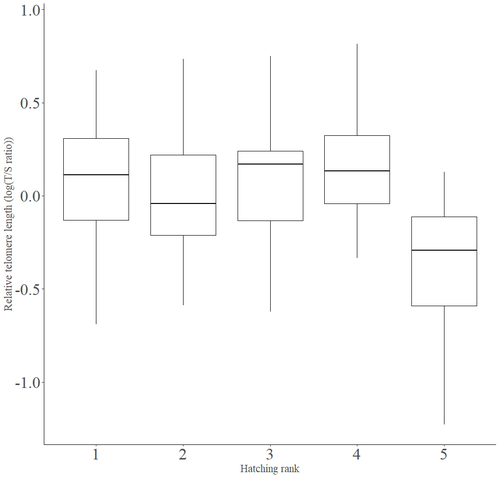
Telomere length vary with sex, hatching rank and year of birth in little owls, Athene noctuaDeciphering the relative contribution of environmental and biological factors driving telomere length in nestlingsRecommended by Jean-François Lemaitre based on reviews by Florentin Remot and 1 anonymous reviewerThe search for physiological markers of health and survival in wild animal populations is attracting a great deal of interest. At present, there is no (and may never be) consensus on such a single, robust marker but of all the proposed physiological markers, telomere length is undoubtedly the most widely studied in the field of evolutionary ecology (Monaghan et al., 2022). Broadly speaking, telomeres are non-coding DNA sequences located at the end of chromosomes in eukaryotes, protecting genomic DNA against oxidative stress and various detrimental processes (e.g. DNA end-joining) and thus maintaining genome stability (Blackburn et al., 2015). However, in most somatic cells from the vast majority of the species, telomere sequences are not replicated and telomere length progressively declines with increased age (Remot et al., 2022). This shortening of telomere length upon a critical level is causally linked to cellular senescence and has been invoked as one of the primary causes of the aging process (López-Otín et al., 2023). Studies performed in both captive and wild populations of animals have further demonstrated that short telomeres (or telomere sequences with a fast attrition rate) are to some extent associated with an increased risk of mortality, even if the magnitude of this association largely differs between species and populations (Wilbourn et al., 2018). The repeated observations of associations between telomere length and mortality risk have called for studies seeking to identify the ecological and biological factors that – beyond chronological age – shape the between-individual variability in telomere length. A wide spectrum of environmental stressors such as the level of exposure to pathogens or the degree of human disturbances has been proposed as possible modulators of telomere dynamics (see Chatelain et al., 2019). However, within species, the relative contribution of various ecological and biological factors on telomere length has been rarely quantified. In that context, the study of Criscuolo and colleagues (2023) constitutes a timely attempt to decipher the relative contribution of environmental and biological factors driving telomere length in nestlings (i.e. when individuals are between 15 and 35 days of age) from a wild population of little owls, Athene noctua. In addition to chronological age, Criscuolo and colleagues (2023) analysed the effects of two environmental variables (i.e. cohort and habitat quality) as well as three life history traits (i.e. hatching rank, sex and body condition). Among these traits, sex was found to impact nestling’s telomere length with females carrying longer telomeres than males. Traditionally, the among-individuals variability in telomere length during the juvenile period is interpreted as a direct consequence of differences in growth allocation. Fast-growing individuals are typically supposed to undergo more cell divisions and a higher exposure to oxidative stress, which ultimately shortens telomeres (Monaghan & Ozanne, 2018). Whether - despite a slightly female-biased sexual size dimorphism - male little owls display a condensed period of fast growth that could explain their shorter telomere is yet to be determined. Future studies should also explore the consequences of these sex differences in telomere length in terms of mortality risk. In birds, it has been observed that telomere length during early life can predict lifespan (see Heidinger et al., 2012 in zebra finches, Taeniopygia guttata), suggesting that females little owls might live longer than their conspecific males. Yet, adult mortality is generally female-biased in birds (Liker & Székely, 2005) and whether little owls constitute an exception to this rule - possibly mediated by sex-specific telomere dynamics - remains to be explored. Quite surprisingly, the present study in little owls did not evidence any clear effect of environmental conditions on nestling’s telomere length, at both temporal and special scales. While a trend for a temporal effect was detected with telomere length being slightly shorter for nestling born the last year of the study (out of 4 years analysed), habitat quality (measured by the proportion of meadow and orchards in the nest environment) had absolutely no impact on nestling telomere length. Recently published studies in wild populations of vertebrates have highlighted the detrimental effects of harsh environmental conditions on telomere length (e.g. Dupoué et al., 2022 in common lizards, Zootoca vivipara), arguing for a key role of telomere dynamics in the emerging field of conservation physiology. While we can recognize the relevance of such an integrative approach, especially in the current context of climate change, the study by Criscuolo and colleagues (2023) reminds us that the relationships between environmental conditions and telomere dynamics are far from straightforward. Depending on the species and its life history, telomere length in early life could indeed capture very different environmental signals. References Blackburn, E. H., Epel, E. S., & Lin, J. (2015). Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science, 350(6265), 1193-1198. https://doi.org/10.1126/science.aab3389 | Telomere length vary with sex, hatching rank and year of birth in little owls, *Athene noctua* | François Criscuolo, Inès Fache, Bertrand Scaar, Sandrine Zahn, Josefa Bleu | <p>Telomeres are non-coding DNA sequences located at the end of linear chromosomes, protecting genome integrity. In numerous taxa, telomeres shorten with age and telomere length (TL) is positively correlated with longevity. Moreover, TL is also af... | | Evolutionary Ecology, Life History | Jean-François Lemaitre | 2023-03-07 09:44:32 | ||
03 Oct 2023
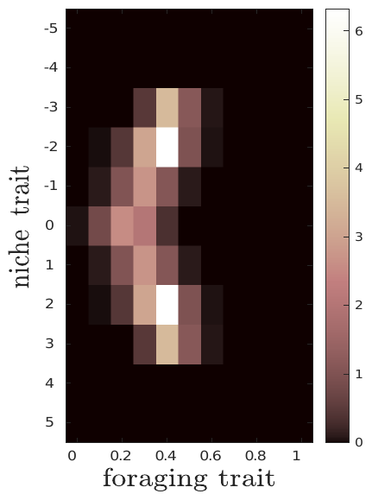
The evolutionary dynamics of plastic foraging and its ecological consequences: a resource-consumer modelEvolution and consequences of plastic foraging behavior in consumer-resource ecosystemsRecommended by François Rousset based on reviews by 2 anonymous reviewersPlastic responses of organisms to their environment may be maladaptive in particular when organisms are exposed to new environments. Phenotypic plasticity may also have opposite effects on the adaptive response of organisms to environmental changes: whether phenotypic plasticity favors or hinders such adaptation depends on a balance between the ability of the population to respond to the change non-genetically in the short term, and the weakened genetic response to environmental change. These topics have received continued attention, particularly in the context of climate change (e.g., Chevin et al. 2013, Duputié et al., 2015, Vinton et al . 2022). In their work, Ledru et al. focus on the adaptive nature of plastic behavior and on its consequences in a consumer-resource ecosystem. As they emphasize, previous works have found that plastic foraging promotes community stability, but these did not consider plasticity as an evolving trait, so Ledru et al. set out to test whether this conclusion holds when both plastic foraging and niche traits of consumers and resources evolve (though ultimately, their new conclusions may not all depend on plasticity evolving). Along the way, they first seek to clarify when such plasticity will evolve, and how it affects the evolution of the niche diversity of consumers and resources, before turning to the question of consumer persistence. The model is rather complex, as three traits are allowed to evolve, and the resource uptake expressed through plastic behavior has its own dynamics affected by some form of social learning. Classically, in models of niche evolution, a consumer's efficiency in exploiting a resource characterized by a trait y (here, the resource's individual niche trait), has been described in terms of location-scale (typically Gaussian) kernels, with mean x (the consumer's individual niche trait) specifying the most efficiently exploited resource, and with variance characterizing individual niche breadth. The evolution of the variance has been considered in some previous models but is assumed to be fixed here. Rather, the new model considers the evolution of the distribution of resource traits, of the consumer's individual niche trait (which is not plastic), and of a "plastic foraging trait" that controls the relative time spent foraging plastically versus foraging randomly. When foraging plastically, the consumers modify their foraging effort towards the type of resource that maximizes their energy intake. in some previous models, the effect of variation in the extent of plastic foraging was already considered, but the evolution of allocation to a plastic foraging strategy versus random foraging was not considered. The model is formulated through reaction-diffusion equations, and its dynamics is investigated by numerical integration. Foraging plasticity readily evolves, when resources vary widely enough, competition for resources is strong, and the cost of plasticity is weak. This means in particular that a large individual niche width of consumers selects for increased plastic foraging, as the evolution of plastic foraging leads to reduced niche overlap between consumers. The evolution of plastic foraging itself generally, though not always, favors the diversification of the niche traits of consumers and of resources. There is thus a positive feedback loop between plastic foraging and resource diversity. Ledru et al. conclude that the total niche width of the consumer population should also correlate with the evolution of plastic foraging, an implication which they relate to the so-called niche variation hypothesis and to empirical tests of it. The joint evolution of the consumer's individual niche trait and plastic foraging trait generates a striking pattern within populations: consumers whose individual niche trait is at an edge of the resource distribution forage more plastically. The authors observe that this relatively simple prediction has not been subjected to any empirical test. Returning to the question of consumer persistence, Ledru et al. evaluate this persistence when consumer mortality increases, and in response to either gradual or sudden environmental changes. These different perturbations all reduce the benefits of plastic foraging. The effect of plastic foraging on stability are complex, being negative or positive effect depending on the type of disturbance, and in particular the ecosystem has a lower sustainable rate of environmental change in the presence of plastic foraging. However, allowing the evolutionary regression of plastic foraging then has a comparatively positive effect on persistence. Despite the substantial effort devoted to analyzing this complex model, relaxing some of its assumptions would likely reveal further complexities. Notably, the overall effect of plasticity on consumer persistence depends on effects already encountered in models of the adaptive response of single species to environmental change: a fast non-genetic response in the short term versus a weakened genetic response in the longer term. The overall balance between these opposite effects on adaptation may be difficult to predict robustly. In the case of a constant rate of environmental change, the results of the present model depend on a lag load between the trait changes of consumer and resource populations, and the extent of the lag may also depend on many factors, such as the extent of genetic variation (e.g., Bürger & Lynch, 1995) for niche traits in consumers and resources. Here, the same variance of mutational effects was assumed for all three evolving traits. Further, spatial environmental variation, a central issue in studies of adaptive responses to environmental changes (e.g., Parmesan, 2006, Zhu et al., 2012), was not considered. Finally, the rate of adjustment of effort by consumers with given niche trait and plastic foraging trait values was assumed proportional to the density of consumers with such trait values. This was justified as a way of accounting for the use of social cues during foraging, but to the extent that they occur, social effects could manifest themselves through other learning dynamics. In conclusion, Ledru et al. have addressed a broad range of questions, suggesting new empirical tests of behavioural patterns on one side, and recovering in the context of community response to environmental changes a complexity that could be expected from earlier works on adaptive responses of organisms but that has been overlooked by previous works on community effects of phenotypic plasticity. References Bürger, R. and Lynch, M. (1995), Evolution and extinction in a changing environment: a quantitative-genetic analysis. Evolution, 49: 151-163. https://doi.org/10.1111/j.1558-5646.1995.tb05967.x Chevin, L.-M., Collins, S. and Lefèvre, F. (2013), Phenotypic plasticity and evolutionary demographic responses to climate change: taking theory out to the field. Funct Ecol, 27: 967-979. https://doi.org/10.1111/j.1365-2435.2012.02043.x Duputié, A., Rutschmann, A., Ronce, O. and Chuine, I. (2015), Phenological plasticity will not help all species adapt to climate change. Glob Change Biol, 21: 3062-3073. https://doi-org.inee.bib.cnrs.fr/10.1111/gcb.12914 Ledru, L., Garnier, J., Guillot, O., Faou, E., & Ibanez, S. (2023). The evolutionary dynamics of plastic foraging and its ecological consequences: a resource-consumer model. EcoEvoRxiv, ver. 4 peer-reviewed and recommended by Peer Community In Evolutionary Biology. https://doi.org/10.32942/X2QG7M Parmesan, C. (2006) Ecological and evolutionary responses to recent climate change Vinton, A.C., Gascoigne, S.J.L., Sepil, I., Salguero-Gómez, R., (2022) Plasticity’s role in adaptive evolution depends on environmental change components. Trends in Ecology & Evolution, 37: 1067-1078. Zhu, K., Woodall, C.W. and Clark, J.S. (2012), Failure to migrate: lack of tree range expansion in response to climate change. Glob Change Biol, 18: 1042-1052. https://doi.org/10.1111/j.1365-2486.2011.02571.x | The evolutionary dynamics of plastic foraging and its ecological consequences: a resource-consumer model | Léo Ledru, Jimmy Garnier, Océane Guillot, Erwan Faou, Camille Noûs, Sébastien Ibanez | <p style="text-align: justify;">Phenotypic plasticity has important ecological and evolutionary consequences. In particular, behavioural phenotypic plasticity such as plastic foraging (PF) by consumers, may enhance community stability. Yet little ... | | Bioinformatics & Computational Biology, Evolutionary Dynamics, Evolutionary Ecology, Phenotypic Plasticity | François Rousset | 2023-03-25 12:04:08 |




