
Latest recommendations
| Id | Title * | Authors * | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
13 Apr 2023
The landscape of nucleotide diversity in Drosophila melanogaster is shaped by mutation rate variationAn unusual suspect: the mutation landscape as a determinant of local variation in nucleotide diversityRecommended by Fernando Racimo based on reviews by David Castellano and 1 anonymous reviewerSometimes, important factors for explaining biological processes fall through the cracks, and it is only through careful modeling that their importance eventually comes out to light. In this study, Barroso and Dutheil introduce a new method based on the sequentially Markovian coalescent (SMC, Marjoran and Wall 2006) for jointly estimating local recombination and coalescent rates along a genome. Unlike previous SMC-based methods, however, their method can also co-estimate local patterns of variation in mutation rates. This is a powerful improvement which allows them to tackle questions about the reasons for the extensive variation in nucleotide diversity across the chromosomes of a species - a problem that has plagued the minds of population geneticists for decades (Begun and Aquadro 1992, Andolfatto 2007, McVicker et al., 2009, Pouyet and Gilbert 2021). The authors find that variation in de novo mutation rates appears to be the most important factor in determining nucleotide diversity in Drosophila melanogaster. Though seemingly contradicting previous attempts at addressing this problem (Comeron 2014), they take care to investigate and explain why that might be the case. Barroso and Dutheil have also taken care to carefully explain the details of their new approach and have carried a very thorough set of analyses comparing competing explanations for patterns of nucleotide variation via causal modeling. The reviewers raised several issues involving choices made by the authors in their analysis of variance partitioning, the proper evaluation of the role of linked selection and the recombination rate estimates emerging from their model. These issues have all been extensively addressed by the authors, and their conclusions seem to remain robust. The study illustrates why the mutation landscape should not be ignored as an important determinant of local variation in genetic diversity, and opens up questions about the generalizability of these results to other organisms. REFERENCES Andolfatto, P. (2007). Hitchhiking effects of recurrent beneficial amino acid substitutions in the Drosophila melanogaster genome. Genome research, 17(12), 1755-1762. https://doi.org/10.1101/gr.6691007 Barroso, G. V., & Dutheil, J. Y. (2021). The landscape of nucleotide diversity in Drosophila melanogaster is shaped by mutation rate variation. bioRxiv, 2021.09.16.460667, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.09.16.460667 Begun, D. J., & Aquadro, C. F. (1992). Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature, 356(6369), 519-520. https://doi.org/10.1038/356519a0 Comeron, J. M. (2014). Background selection as baseline for nucleotide variation across the Drosophila genome. PLoS Genetics, 10(6), e1004434. https://doi.org/10.1371/journal.pgen.1004434 Marjoram, P., & Wall, J. D. (2006). Fast" coalescent" simulation. BMC genetics, 7, 1-9. https://doi.org/10.1186/1471-2156-7-16 McVicker, G., Gordon, D., Davis, C., & Green, P. (2009). Widespread genomic signatures of natural selection in hominid evolution. PLoS genetics, 5(5), e1000471. https://doi.org/10.1371/journal.pgen.1000471 Pouyet, F., & Gilbert, K. J. (2021). Towards an improved understanding of molecular evolution: the relative roles of selection, drift, and everything in between. Peer Community Journal, 1, e27. https://doi.org/10.24072/pcjournal.16 | The landscape of nucleotide diversity in Drosophila melanogaster is shaped by mutation rate variation | Gustavo V Barroso, Julien Y Dutheil | <p style="text-align: justify;">What shapes the distribution of nucleotide diversity along the genome? Attempts to answer this question have sparked debate about the roles of neutral stochastic processes and natural selection in molecular evolutio... | | Bioinformatics & Computational Biology, Population Genetics / Genomics | Fernando Racimo | 2022-10-30 07:52:07 | ||
11 Apr 2023

Facultative parthenogenesis: a transient state in transitions between sex and obligate asexuality in stick insects?Facultative parthenogenesis and transitions from sexual to asexual reproductionRecommended by Trine Bilde based on reviews by 3 anonymous reviewers based on reviews by 3 anonymous reviewers
Despite a vast array of ways in which organisms can reproduce (Bell, 1982), most animals engage in sexual reproduction (Otto & Lenormand, 2002). A fascinating alternative to sex is parthenogenesis, where offspring are produced asexually from a gamete, typically the egg, without receiving genetic material from another gamete (Simon, Delmotte, Rispe, & Crease, 2003). One of the long-standing questions in the field is why parthenogenesis is not more widespread, given the costs associated with sex (Otto & Lenormand, 2002). Natural populations of most species appear to be reproducing either sexually or parthenogenetically, even if a species can employ both reproductive modes (Larose et al 2023). Larose et al (2023) highlight the conundrum in this pattern, as organisms that are capable of employing parthenogenesis facultatively would be able to gain the benefits of both modes of reproduction. Why then, is facultative parthenogenesis not more common? Larose et al (2023) propose that constraints on being efficient in both sexual and asexual reproduction could cause a trade-off between reproductive modes that favours an obligate strategy of either sex or no sex. This would provide an explanation for why facultative parthenogenesis is rare. Timema stick insects provide an excellent system to investigate reproductive strategies, as some species have parthenogenetic females, while other species are sexual, and they show repeated transitions from sexual reproduction to obligate parthenogenesis (Schwander & Crespi, 2009). The authors performed comprehensive and complementary studies in a recently discovered species T. douglasi, in which populations show both modes of reproduction, with some populations consisting only of females and others showing equal proportions of males and females. The sex ratio varied significantly, with the proportion of females ranging between 43-100% across 29 populations. These populations form a monophyletic clade with clustering into three genetic lineages and only a few cases of admixture. Females from all populations were capable of producing unfertilized eggs, but the hatching success varied hugely among populations and lineages (3-100%). Parthenogenetically produced offspring were homozygous, showing that parthenogenesis causes a complete loss of heterozygosity in a single generation. After producing eggs as virgins, females were mated to assess the capacity to also reproduce sexually, and fertilization increased the hatching success of eggs in two lineages. In one lineage, in which the hatching success of unfertilized eggs is similar to that of other sexually reproducing Timema species, fertilization reduced egg-hatching success, indicating a trade-off between reproductive modes with parthenogenetic reproduction performing best. Approximately 58% of the offspring produced after mating were fertilized, demonstrating the capacity of females to reproduce parthenogenetically also after mating has occurred, however with huge variation among individuals. This wonderful and meticulously performed study produces strong and complementary evidence for facultative parthenogenesis in T. douglasi populations. The study shows large variation in how reproductive mode is employed, supporting the existence of a trade-off between sexual and parthenogenetic reproduction. This might be an example of an ongoing transition from sexual to asexual reproduction, which indicates that obligate parthenogenesis may derive via transient facultative parthenogenesis. REFERENCES Bell, G. (1982) The Masterpiece of Nature: The Evolution and Genetics of Sexuality. University of California Press. 635 p. Otto, S. P., & Lenormand, T. (2002). Resolving the paradox of sex and recombination. Nature Reviews Genetics, 3(4), 252-261. https://doi.org/10.1038/nrg761 Schwander, T., & Crespi, B. J. (2009). Multiple direct transitions from sexual reproduction to apomictic parthenogenesis in Timema stick insects. Evolution, 63(1), 84-103. Simon, J.-C., Delmotte, F., Rispe, C., & Crease, T. (2003). Phylogenetic relationships between parthenogens and their sexual relatives: the possible routes to parthenogenesis in animals. Biological Journal of the Linnean Society, 79(1), 151-163. https://doi.org/10.1046/j.1095-8312.2003.00175.x Larose, C., Lavanchy, G., Freitas, S., Parker, D.J., Schwander, T. (2023) Facultative parthenogenesis: a transient state in transitions between sex and obligate asexuality in stick insects? bioRxiv, 2022.03.25.485836, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.03.25.485836 | Facultative parthenogenesis: a transient state in transitions between sex and obligate asexuality in stick insects? | Chloé Larose, Guillaume Lavanchy, Susana Freitas, Darren J. Parker, Tanja Schwander | <p>Transitions from obligate sex to obligate parthenogenesis have occurred repeatedly across the tree of life. Whether these transitions occur abruptly or via a transient phase of facultative parthenogenesis is rarely known. We discovered and char... | | Reproduction and Sex | Trine Bilde | 2022-05-20 10:41:13 | ||
30 Mar 2023
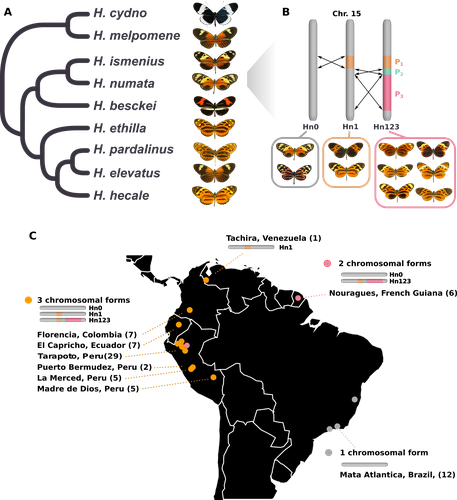
Balancing selection at a wing pattern locus is associated with major shifts in genome-wide patterns of diversity and gene flow in a butterflyIs genetic diversity enhanced by a supergene?Recommended by Chris Jiggins based on reviews by Christelle Fraïsse and 2 anonymous reviewersThe butterfly species Heliconius numata has a remarkable wing pattern polymorphism, with multiple pattern morphs all controlled by a single genetic locus, which harbours multiple inversions. Each morph is a near-perfect mimic of a species in the fairly distantly related genus of butterflies, Melinaea. The article by Rodríguez de Cara et al (2023) argues that the balanced polymorphism at this single wing patterning locus actually has a major effect on genetic diversity across the whole genome. First, polymorphic populations within H. numata are more dioverse than those without polymorphism. Second, H. numata is more genetically diverse than other related species and finally reconstruction of historical demography suggests that there has been a recent increase in effective population size, putatively associated with the acquisition of the supergene polymorphism. The supergene itself generates disassortative mating, such that morphs prefer to mate with others dissimilar to themselves - in this way it is similar to mechanisms for preventing inbreeding such as self-incompatibility loci in plants. This provides a potential mechanism whereby non-random mating patterns could increase effective population size. The authors also explore this mechanism using forward simulations, and show that mating patterns at a single locus can influence linked genetic diversity over a large scale. Overall, this is an intriguing study, which suggests a far more widespread genetic impact of a single locus than might be expected. There are interesting parallels with mechanisms of inbreeding prevention in plants, such as the Pin/Thrum polymorphism in Primula, which also rely on mating patterns determined by a single locus but presumably also influence genetic diversity genome-wide by promoting outbreeding. REFERENCES Rodríguez de Cara MÁ, Jay P, Rougemont Q, Chouteau M, Whibley A, Huber B, Piron-Prunier F, Ramos RR, Freitas AVL, Salazar C, Silva-Brandão KL, Torres TT, Joron M (2023) Balancing selection at a wing pattern locus is associated with major shifts in genome-wide patterns of diversity and gene flow. bioRxiv, 2021.09.29.462348, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.09.29.462348 | Balancing selection at a wing pattern locus is associated with major shifts in genome-wide patterns of diversity and gene flow in a butterfly | María Ángeles Rodríguez de Cara, Paul Jay, Quentin Rougemont, Mathieu Chouteau, Annabel Whibley, Barbara Huber, Florence Piron-Prunier, Renato Rogner Ramos, André V. L. Freitas, Camilo Salazar, Karina Lucas Silva-Brandão, Tatiana Texeira Torres, M... | <p style="text-align: justify;">Selection shapes genetic diversity around target mutations, yet little is known about how selection on specific loci affects the genetic trajectories of populations, including their genomewide patterns of diversity ... | | Evolutionary Ecology, Genome Evolution, Hybridization / Introgression, Population Genetics / Genomics | Chris Jiggins | 2021-10-13 17:54:33 | ||
24 Mar 2023
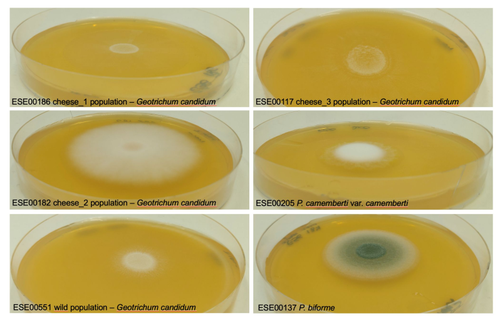
Domestication of different varieties in the cheese-making fungus Geotrichum candidumDiverse outcomes in cheese fungi domesticationRecommended by Christelle Fraïsse based on reviews by Delphine Sicard and 1 anonymous reviewer
Domestication is a complex process that imprints the demography and the genomes of domesticated populations, enforcing strong selective pressures on traits favourable to humans, e.g. for food production [1]. Domestication has been quite intensely studied in plants and animals, but less so in micro-organisms such as fungi, despite their assets (e.g. their small genomes and tractability in the lab). This elegant study by Bennetot and collaborators [2] on the cheese-making fungus Geotrichum candidum adds to the mounting body of studies in the genomics of fungi, proving they are excellent models in evolutionary biology for studying adaptation and drift in eukaryotes [3]. Bennetot et al. newly showed with whole genome sequences that all G. candidum strains isolated from cheese form a monophyletic clade subdivided into three genetically differentiated populations with several admixed strains, while the wild strains sampled from diverse geographic locations form a sister clade. This suggests the wild progenitor was not sampled in the present study and calls for future exciting work on the domestication history of the G. candidum fungus. The authors scanned the genomes for footprints of adaptation to the cheese environment and identified promising candidates, such as a gene involved in iron uptake (this element is limiting in cheese). Their functional genome analysis also provides evidence for higher contents of transposable elements in cheese-making strains, likely due to relaxed selection during the domestication process. This paper is particularly impressive in that the authors complemented the population genomic approach with the phenotypic characterization of the strains and tested their ability to outcompete common fungal food spoilers. The authors convincingly showed that cheese-making strains display phenotypic differences relative to wild relatives for multiple traits such as slower growth, lower proteolysis activity and a greater amount of volatiles attractive to consumers, these phenotypes being beneficial for cheese making. Finally, this work is particularly inspiring because it thoroughly discusses convergent evolution during domestication in different cheese-associated fungi. Indeed, studying populations experiencing similar environmental pressures is fundamental to understanding whether evolution is repeatable [4]. For instance, all three cheese populations of G. candidum exhibit a lower genetic diversity than wild populations. However, only one population displays a stronger domestication syndrome, resembling the Penicillium camemberti situation [5]. Furthermore, different cheese-making practices may have led to varying situations with clonal lineages in non-Roquefort P. roqueforti and P. camemberti [5, 6], while the cheese-making G. candidum populations still harbour some diversity. In a nutshell, Bennetot's study makes an important contribution to evolutionary biology and highlights the value of diversifying our model organisms toward under-represented clades. REFERENCES [1] Diamond J (2002) Evolution, consequences and future of plant and animal domestication. Nature 418: 700–707. https://doi.org/10.1038/nature01019 [2] Bennetot B, Vernadet J-P, Perkins V, Hautefeuille S, Rodríguez de la Vega RC, O’Donnell S, Snirc A, Grondin C, Lessard M-H, Peron A-C, Labrie S, Landaud S, Giraud T, Ropars J (2023) Domestication of different varieties in the cheese-making fungus Geotrichum candidum. bioRxiv, 2022.05.17.492043, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.05.17.492043 [3] Gladieux P, Ropars J, Badouin H, Branca A, Aguileta G, de Vienne DM, Rodríguez de la Vega RC, Branco S, Giraud T (2014) Fungal evolutionary genomics provides insight into the mechanisms of adaptive divergence in eukaryotes. Mol. Ecol. 23: 753–773. https://doi.org/10.1111/mec.12631 [4] Bolnick DI, Barrett RD, Oke KB, Rennison DJ, Stuart YE (2018) (Non)Parallel evolution. Ann. Rev. Ecol. Evol. Syst. 49: 303–330. https://doi.org/10.1146/annurev-ecolsys-110617-062240 [5] Ropars J, Didiot E, Rodríguez de la Vega RC, Bennetot B, Coton M, Poirier E, Coton E, Snirc A, Le Prieur S, Giraud T (2020) Domestication of the Emblematic White Cheese-Making Fungus Penicillium camemberti and Its Diversification into Two Varieties. Current Biol. 30: 4441–4453.e4. https://doi.org/10.1016/j.cub.2020.08.082 [6] Dumas, E, Feurtey, A, Rodríguez de la Vega, RC, Le Prieur S, Snirc A, Coton M, Thierry A, Coton E, Le Piver M, Roueyre D, Ropars J, Branca A, Giraud T (2020) Independent domestication events in the blue-cheese fungus Penicillium roqueforti. Mol Ecol. 29: 2639–2660. https://doi.org/10.1111/mec.15359 | Domestication of different varieties in the cheese-making fungus *Geotrichum candidum* | Bastien Bennetot, Jean-Philippe Vernadet, Vincent Perkins, Sophie Hautefeuille, Ricardo C. Rodríguez de la Vega, Samuel O’Donnell, Alodie Snirc, Cécile Grondin, Marie-Hélène Lessard, Anne-Claire Peron, Steve Labrie, Sophie Landaud, Tatiana Giraud,... | <p>Domestication is an excellent model for studying adaptation processes, involving recent adaptation and diversification, convergence following adaptation to similar conditions, as well as degeneration of unused functions. <em>Geotrichum candidum... | | Adaptation, Genome Evolution, Population Genetics / Genomics | Christelle Fraïsse | 2022-08-12 20:50:42 | ||
16 Mar 2023
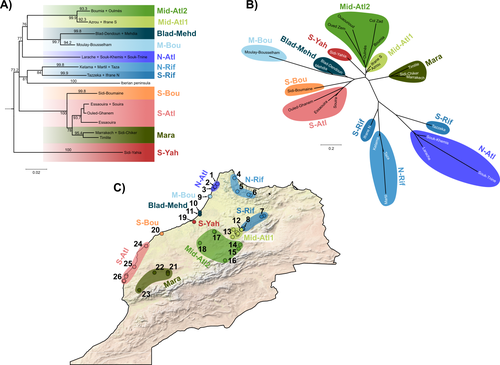
Phylogeographic breaks and how to find them: Separating vicariance from isolation by distance in a lizard with restricted dispersalThe difficult task of partitioning the effects of vicariance and isolation by distance in poor dispersersRecommended by Eric Pante based on reviews by Kevin Sánchez and Aglaia (Cilia) Antoniou
Partitioning the effects of vicariance and low dispersal has been a long-standing problem in historical biogeography and phylogeography. While the term “vicariance” refers to divergence in allopatry, caused by some physical (geological, geographical) or climatic barriers (e.g. Rosen 1978), isolation by distance refers to the genetic differentiation of remote populations due to the physical distance separating them, when the latter surpasses the scale of dispersal (Wright 1938, 1940, 1943). Vicariance and dispersal have long been considered as separate forces leading to separate scenarii of speciation (e.g. reviewed in Hickerson and Meyer 2008). Nevertheless, these two processes are strongly linked, as, for example, vicariance theory relies on the assumption that ancestral lineages were once linked by dispersal prior to physical or climatic isolation (Rosen 1978). Low dispersal and vicariance are not mutually exclusive, and distinguishing these two processes in heterogeneous landscapes, especially for poor dispersers, remains therefore a severe challenge. For example, low dispersal (and/or small population size) can give rise to geographic patterns consistent with a phylogeographic break and be mistaken for geographic isolation (Irwin 2002, Kuo and Avise 2005). The study of Rancilliac and colleagues (2023) is at the heart of this issue. It focuses on a nominal lizard species, the red-tailed spiny-footed lizard (Acanthodactylus erythrurus, Squamata: Lacertidae), which has a wide spatial distribution (from the Maghreb to the Iberian Peninsula), is found in a variety of different habitats, and has a wide range of morphological traits that do not always correlate with phylogeny. The main question is the following: have “the morphological and ecological diversification of this group been produced by vicariance and lineage diversification, or by local adaptation in the face of historical gene flow?” To tackle this question, the authors used sequence data from multiple mitochondrial and nuclear markers and a nested analysis workflow integrating phylogeography, multiple correspondence analyses and a relatively novel approach to IBD testing (Hausdorf & Henning, 2020). The latter is based on regression analysis and was shown to be less prone to error than the traditional (partial) Mantel test. While this set of methods allowed the partitioning of the effect of isolation by distance and vicariance in shaping contemporary genetic diversity in red-tailed spiny-footed lizards, some of the evolutionary history of this species complex remains blurred by ongoing gene flow and admixture, retention of ancestral polymorphism, or selection. The lack of congruence between mitochondrial and nuclear gene trees once again warns us that proposing evolutionary scenarii based on individual gene trees is a risky business. References Hausdorf B, Hennig C (2020) Species delimitation and geography. Molecular Ecology Resources, 20, 950–960. https://doi.org/10.1111/1755-0998.13184 Hickerson MJ, Meyer CP (2008) Testing comparative phylogeographic models of marine vicariance and dispersal using a hierarchical Bayesian approach. BMC Evolutionary Biology, 8, 322. https://doi.org/10.1186/1471-2148-8-322 Irwin DE (2002) Phylogeographic breaks without geographic barriers to gene flow. Evolution, 56, 2383–2394. https://doi.org/10.1111/j.0014-3820.2002.tb00164.x Kuo C-H, Avise JC (2005) Phylogeographic breaks in low-dispersal species: the emergence of concordance across gene trees. Genetica, 124, 179–186. https://doi.org/10.1007/s10709-005-2095-y Rancilhac L, Miralles A, Geniez P, Mendez-Aranda D, Beddek M, Brito JC, Leblois R, Crochet P-A (2023) Phylogeographic breaks and how to find them: An empirical attempt at separating vicariance from isolation by distance in a lizard with restricted dispersal. bioRxiv, 2022.09.30.510256, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.09.30.510256 Rosen DE (1978) Vicariant Patterns and Historical Explanation in Biogeography. Systematic Biology, 27, 159–188. https://doi.org/10.2307/2412970 Wright, S (1938) Size of population and breeding structure in relation to evolution. Science 87:430-431. Wright S (1940) Breeding Structure of Populations in Relation to Speciation. The American Naturalist, 74, 232–248. https://doi.org/10.1086/280891 Wright S (1943) Isolation by distance. Genetics, 28, 114–138. https://doi.org/10.1093/genetics/28.2.114 | Phylogeographic breaks and how to find them: Separating vicariance from isolation by distance in a lizard with restricted dispersal | Loïs Rancilhac, Aurélien Miralles, Philippe Geniez, Daniel Mendez-Arranda, Menad Beddek, José Carlos Brito, Raphaël Leblois, Pierre-André Crochet | <p>Aim</p> <p>Discontinuity in the distribution of genetic diversity (often based on mtDNA) is usually interpreted as evidence for phylogeographic breaks, underlying vicariant units. However, a misleading signal of phylogeographic break can arise... | | Phylogeography & Biogeography, Population Genetics / Genomics, Speciation, Systematics / Taxonomy | Eric Pante | Kevin Sánchez | 2022-10-05 13:11:28 | |
06 Mar 2023
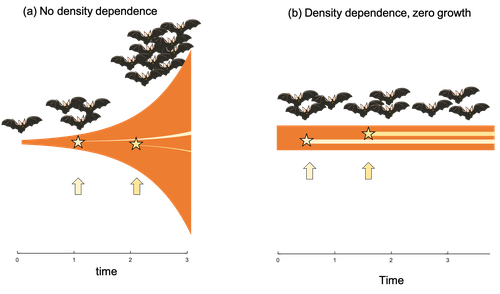
Extrinsic mortality and senescence: a guide for the perplexedGetting old gracefully, and risk of dying before getting there: a new guide to theory on extrinsic mortality and senescenceRecommended by Sinead English and Shinichi Nakagawa based on reviews by Nicole Walasek and 1 anonymous reviewerWhy is there such variation across species and populations in the rate at which individuals show wear and tear as they get older? Several compelling theoretical explanations have been developed on the conditions under which selection allows for or prevents senescence; a notable one being that proposed by George C Williams in 1957 based on the idea of antagonistic pleiotropy (Williams, 1957). One of the testable predictions of this theory is that, in populations where adults experience higher mortality, senescence is expected to be faster. This is one of the most influential predictions of the paper, being intuitive (when individuals are less likely to survive to later age classes, we expect weakened selection on traits that would avoid senescence in these classes), and fitting with ‘live fast, die young’ life history framing. As such, it has been widely incorporated into how we think about the evolution of senescence and has received considerable support in comparative studies across species and populations. However, it would be misleading to sit back at this point and think we have ‘solved’ the problem of understanding variation in senescence, and how this is linked with mortality. It turns out that the Williams 1957 paper is hotly contested by theoreticians: for the past 30 years – with increasing focus in the last 4 years – a growing body of models and opinion pieces have proposed flaws in the paper itself and in how it has been interpreted (Abrams, 1993; André and Rousset, 2020; Day and Abrams, 2020; Moorad et al., 2019). Central to several of these critiques is that explicit consideration of density dependence (not considered in Williams’ original paper) changes the conditions under which his predictions hold. A new preprint by de Vries, Gallipaud and Kokko brings further clarity to such critiques of the original paper (Vries et al., 2023). Beyond just continuing the tradition of critiquing Williams’ prediction, however, de Vries et al. provide a clear guide that is accessible to non-theoreticians about the issues with William’s prediction, and the way in which density dependence and how it operates can change when we expect senescence to occur. Rather than reiterate their points here, we suggest a close reading of the paper itself, along with an excellent overview of the paper in a recent blog by Daniel Nettle (Nettle, 2022). In brief, the paper starts by synthesizing earlier theoretical and empirical studies on the topic and presenting a very simple model to highlight how – in the absence of density dependence – Williams’ prediction does not hold because of the unregulated population growth, which is necessarily higher when there is low mortality. As a result, for a lineage with low mortality, any fitness advantage of placing offspring into the lineage later (i.e. selection for reduced senescence) is exactly cancelled out by the fact that earlier-produced offspring have higher fitness returns. They then present a more complex framework, which incorporates realistic mortality distributions, trade-offs between survival and reproduction, and use a series of 10 scenarios of density dependence (and whether this acts on survival or fecundity, and also whether it depends on a threshold or stochastic, or exerts continuing pressure on the trait) to explore selection on fitness-associated traits with age depending on extrinsic mortality. This then generates a range of results for when the Williams prediction holds, when there is no link between mortality and senescence, and when there is an ‘anti-Williams’ result – i.e., where senescence is slower when there is a high mortality. As has been shown in earlier studies, density dependence and how it operates matters, and Williams’ prediction holds most when density dependence affects juvenile age classes (in their model, when adults are less likely to produce them – i.e. there is density dependence on fecundity; or when there is less recruitment into the adult population due to, for example, competition among juveniles). So, perhaps we are now at a point where we can lay to rest the debate on the issues specifically with Williams’ original paper, and instead consider more broadly the key factors to measure when predicting patterns of senescence, and what is tangible for empiricists to quantify in their studies. Here, de Vries et al. provide some helpful insights both into the limitations of their approach and what modelling should be done moving forward, and into how we can link existing studies (for example comparing senescence among populations with varying predation pressure) to the theoretical predictions. At the heart of the latter is understanding the mechanism of density-dependent regulation – does it affect survival or fecundity, which age classes are most sensitive, and how do key traits depend on density? – and this is often difficult to measure in field studies. And from all this we can learn that even very intuitive and extensively discussed concepts in biology do not always have as firm theoretical underpinnings as assumed, and – as should not be surprising – biology is complex and rather than one clear pattern being predicted, this can depend on a multitude of factors. REFERENCES Abrams PA (1993) Does increased mortality favor the evolution of more rapid senescence? Evolution, 47, 877–887. https://doi.org/10.1111/j.1558-5646.1993.tb01241.x André J-B, Rousset F (2020) Does extrinsic mortality accelerate the pace of life? A bare-bones approach. Evolution and Human Behavior, 41, 486–492. https://doi.org/10.1016/j.evolhumbehav.2020.03.002 Day T, Abrams PA (2020) Density Dependence, Senescence, and Williams’ Hypothesis. Trends in Ecology & Evolution, 35, 300–302. https://doi.org/10.1016/j.tree.2019.11.005 Moorad J, Promislow D, Silvertown J (2019) Evolutionary Ecology of Senescence and a Reassessment of Williams’ ‘Extrinsic Mortality’ Hypothesis. Trends in Ecology & Evolution, 34, 519–530. https://doi.org/10.1016/j.tree.2019.02.006 Nettle AD (2022) Live fast and die young (maybe). https://www.danielnettle.org.uk/2022/02/18/live-fast-and-die-young-maybe/ (accessed 2.27.23). de Vries C, Galipaud M, Kokko H (2023) Extrinsic mortality and senescence: a guide for the perplexed. bioRxiv, 2022.01.27.478060, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.01.27.478060 Williams GC (1957) Pleiotropy, natural selection, and the evolution of senescence. Evolution, 11, 398–411. https://doi.org/10.1111/j.1558-5646.1957.tb02911.x | Extrinsic mortality and senescence: a guide for the perplexed | Charlotte de Vries, Matthias Galipaud, Hanna Kokko | <p style="text-align: justify;">Do environments or species traits that lower the mortality of individuals create selection for delaying senescence? Reading the literature creates an impression that mathematically oriented biologists cannot agree o... | | Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Theory, Life History | Sinead English | 2022-08-26 14:30:16 | ||
28 Feb 2023
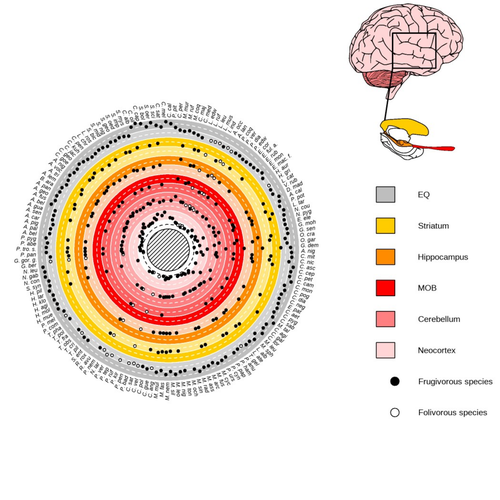
Primate sympatry shapes the evolution of their brain architectureMacroevolutionary drivers of brain evolution in primatesRecommended by Fabien Condamine based on reviews by Paula Gonzalez, Orlin Todorov and 3 anonymous reviewersStudying the evolution of animal cognition is challenging because many environmental and species-related factors can be intertwined, which is further complicated when looking at deep-time evolution. Previous knowledge has emphasized the role of intraspecific interactions in affecting the socio-ecological environment shaping cognition. However, much less is known about such an effect at the interspecific level. Yet, the coexistence of different species in the same geographic area at a given time (sympatry) can impact the evolutionary history of species through character displacement due to biotic interactions. Trait evolution has been observed and tested with morphological external traits but more rarely with brain evolution. Compared to most species’ traits, brain evolution is even more delicate to assess since specific brain regions can be involved in different functions, may they be individual-based and social-based information processing. In a very original and thoroughly executed study, Robira & Perez-Lamarque (2023) addressed the question: How does the co-occurrence of congeneric species shape brain evolution and influence species diversification? By considering brain size as a proxy for cognition, they evaluated whether species sympatry impacted the evolution of cognition in frugivorous primates. Fruit resources are hard to find, not continuous through time, heterogeneously distributed across space, but can be predictable. Hence, cognition considerably shapes the foraging strategy and competition for food access can be fierce. Over long timescales, it remains unclear whether brain size and the pace of species diversification are linked in the context of sympatry, and if so how. Recent studies have found that larger brain sizes can be associated with higher diversification rates in birds (Sayol et al. 2019). Similarly, Robira & Perez-Lamarque (2023) thus wondered if the evolution of brain size in primates impacted their dynamic of species diversification, which has been suggested (Melchionna et al. 2020) but not tested. Prior to anything, Robira & Perez-Lamarque (2023) had to retrace the evolutionary history of sympatry between frugivorous primate lineages through time using the primate tree of life, species’ extant distribution, and process-based models to estimate ancestral range evolution. To infer the effect of species sympatry on the evolution of cognition in frugivorous primates, the authors evaluated the support for phylogenetic models of brain size evolution accounting or not for species sympatry and investigated the directionality of the selection induced by sympatry on brain size evolution. Finally, to better understand the impact of cognition and interactions between primates on their evolutionary success, they tested for correlations between brain size or species’ sympatry and species diversification. Robira & Perez-Lamarque (2023) found that the evolution of the whole brain or brain regions used in immediate information processing was best fitted with models not considering sympatry. By contrast, models considering species sympatry best predicted the evolution of brain regions related to long-term memory of interactions with the socio-ecological environment, with a decrease in their size along with stronger sympatry. Specifically, they found that sympatry was associated with a decrease in the relative size of the hippocampus and striatum, but had no significant effect on the neocortex, cerebellum, or overall brain size. The hippocampus is a brain region that plays a crucial role in processing and memorizing spatiotemporal information, which is relevant for frugivorous primates in their foraging behavior. The study suggests that competition between sympatric species for limited food resources may lead to a more complex and unpredictable food distribution, which may in turn render cognitive foraging not advantageous and result in a selection for smaller brain regions involved in foraging. Niche partitioning and dietary specialization in sympatry may also impact cognitive abilities, with more specialized diets requiring lower cognitive abilities and smaller brain region sizes. On the other hand, the absence of an effect of sympatry on brain regions involved in immediate sensory information processing, such as the cerebellum and neocortex, suggests that foragers do not exploit cues left out by sympatric heterospecific species, or they may discard environmental cues in favor of social cues. This is a remarkable study that highlights the importance of considering the impact of ecological factors, such as sympatry, on the evolution of specific brain regions involved in cognitive processes, and the potential trade-offs in brain region size due to niche partitioning and dietary specialization in sympatry. Further research is needed to explore the mechanisms behind these effects and to test for the possible role of social cues in the evolution of brain regions. This study provides insights into the selective pressures that shape brain evolution in primates. References Melchionna M, Mondanaro A, Serio C, Castiglione S, Di Febbraro M, Rook L, Diniz-Filho JAF, Manzi G, Profico A, Sansalone G, Raia P (2020) Macroevolutionary trends of brain mass in Primates. Biological Journal of the Linnean Society, 129, 14–25. https://doi.org/10.1093/biolinnean/blz161 Robira B, Perez-Lamarque B (2023) Primate sympatry shapes the evolution of their brain architecture. bioRxiv, 2022.05.09.490912, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.05.09.490912 Sayol F, Lapiedra O, Ducatez S, Sol D (2019) Larger brains spur species diversification in birds. Evolution, 73, 2085–2093. https://doi.org/10.1111/evo.13811 | Primate sympatry shapes the evolution of their brain architecture | Benjamin Robira, Benoit Perez-Lamarque | <p style="text-align: justify;">The main hypotheses on the evolution of animal cognition emphasise the role of conspecifics in affecting the socio-ecological environment shaping cognition. Yet, space is often simultaneously occupied by multiple sp... | | Behavior & Social Evolution, Bioinformatics & Computational Biology, Evolutionary Ecology, Macroevolution, Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Fabien Condamine | 2022-05-10 13:43:02 | ||
22 Feb 2023
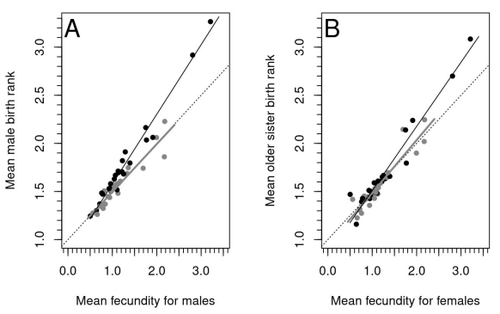
Increased birth rank of homosexual males: disentangling the older brother effect and sexual antagonism hypothesisEvolutionary or proximal explanations for human male homosexual mate preference?Recommended by Jacqui A. Shykoff based on reviews by Ray Blanchard and 1 anonymous reviewer
Natural populations do not consist of only perfectly adapted individuals. If they did, of course, there would be no fodder for evolution by natural selection. And natural selection is operating all the time, winnowing out less well adapted phenotypes through differential reproduction and survival. Demonstrations of natural selection modifying characters-state distributions to bring phenotypes closer to their optima abound in the evolution literature, with examples of short- and long-term changes in phenotype and allele frequencies. However, evolutionary biologists know that populations cannot reach their adaptive peaks. Natural selection is tracking a moving target, always with some generations of lag time. The adaptive landscape is multidimensional, so the optimal combination of multiple character states may be impossible because of constraints and trade-offs. Natural selection does not operate alone or in isolation – new mutations and migrants that were selected under other conditions will inject locally non-adaptive genetic variation and genetic drift can change allele frequencies in random directions. We understand these processes that generate and maintain less advantageous variants on a continuous gradient from an optimal phenotype in a fitness landscape. More puzzling are heritable polymorphisms with distinct morphologies, physiologies or behaviours maintained in populations despite their measurably lower reproductive success. But a complete model of evolution must also be able to accommodate these Darwinian paradoxes. Raymond et al. (2023) investigate one such Darwinian paradox: In humans, male homosexual mate preference is heritable and is associated with a large reduction in offspring production but nonetheless occurs at relatively high frequencies in most human populations. Furthermore, multiple studies have found that homosexual men come from families that are, on average, larger than those of heterosexual men and that homosexual men have, on average, higher birth rank than do heterosexual men, i.e., having more older siblings and, particularly, more older brothers. Two types of mechanisms consistent with these observations have been proposed: 1) An evolutionary mechanism of sex-antagonistic pleiotropy, whereby highly fecund mothers are more likely to produce homosexual sons, and 2) A mechanistic explanation whereby successive male pregnancies alter the uterine environment by increasing the probability of an immune reaction by the mother to her male fetus, altering development of sexually dimorphic brain structures relevant to sexual orientation. In this article, the authors explore these two mechanisms of sex-antagonistic effects (AE) and fraternal birth order effects (FBOE) and test how well they account for patterns of male homosexuality in population and family data. Clearly, these two effects are somewhat confounded because high birth ranks can only occur in large families. If, indeed, the probability of male homosexuality increases with increasing numbers of (maternal) older brothers, homosexual males will be more common in larger families. Similarly, if high female fecundity leads to a higher probability of male homosexuality via sex-antagonistic effects, homosexual males will, on average, have more older brothers. To disentangle the actions of these two effects the authors modelled the relationship between birth rank and population fecundity and investigated whether AE or FBOE modified this relationship for homosexual men. Simulation results were compared with aggregated population data from 13 countries. Family data on individuals’ sexual preference, birth rank and number of male and female siblings from France, Greece and Indonesia were analysed with generalised linear models and Bayesian approaches to test for a signal of AE or FBOE. These analyses revealed a significant older-brother effect (FBOE) explaining patterns of occurrence of homosexuality in population and family data but no significant independent sex-antagonistic effect (AE). Thus larger family sizes of homosexual men appear due to the older-brother effect, with individuals of high birth rank coming necessarily from large sibships. The simulation approach also revealed that modelling a fraternal birth order effect (FBOE), such that individuals with more older brothers are more likely to be homosexual, generates an artefactual older sister effect simply because homosexual men are overrepresented at higher birth ranks. Older-sister effects reported in the literature may, therefore, be statistical artefacts of an underlying older-brother effect. This paper is interesting for a number of reasons. It does an excellent job of explaining, identifying and dealing with estimation biases and testing for artefactual relationships generated by collinearity. It applies state-of-the art analytical/statistical tools. It breaks down two colinear effects and shows that only one really explains phenotypic variation. This is a great example of how to disentangle correlated variables that may or may not both contribute to trait variation. But most intriguingly, we are left without evidence for an evolutionary mechanism that compensates the large fitness cost associated with male homosexuality in humans. How can we explain high heritability maintained in the face of strong directional selection that should erode heritable genetic variation? The usual suspects include cryptic compensatory mechanisms yet to be discovered or flawed estimates of selection or heritability. For example, data on heritability of male homosexual mate preference in humans come from twin studies and twins share birth rank as well as alleles. Thus it is possible that heritability is over-estimated, including the environmental component associated with birth rank. If, as the authors demonstrate here, birth rank is the strongest predictor of male homosexual mate preference, selection may be acting on a non-heritable plastic component of phenotypic variation. This could explain why heritable variation is not exhausted by selection, rendering the paradox less paradoxical, but fails to provide an adaptive explanation for the maintenance of male homosexual mate preference. References Raymond M., Turek D., Durand V., Nila S., Suryobroto B., Vadez J., Barthes J., Apostolou M. and Crochet P.-A. (2023) Increased birth rank of homosexual males: disentangling the older brother effect and sexual antagonism hypothesis. bioRxiv, 2022.02.22.481477, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.02.22.481477 | Increased birth rank of homosexual males: disentangling the older brother effect and sexual antagonism hypothesis | Michel Raymond, Daniel Turek, Valerie Durand, Sarah Nila, Bambang Suryobroto, Julien Vadez, Julien Barthes, Menelaos Apostolou, Pierre-André Crochet | <p style="text-align: justify;">Male homosexual orientation remains a Darwinian paradox, as there is no consensus on its evolutionary (ultimate) determinants. One intriguing feature of homosexual men is their higher male birth rank compared to het... | | Life History, Other, Phenotypic Plasticity, Reproduction and Sex | Jacqui A. Shykoff | 2022-03-03 11:28:44 | ||
21 Feb 2023
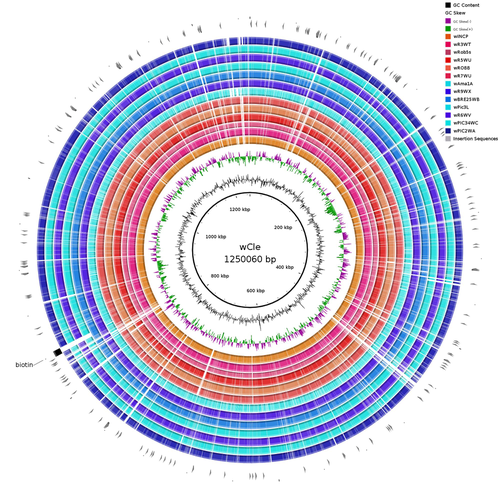
Wolbachia genomics reveals a potential for a nutrition-based symbiosis in blood-sucking Triatomine bugsNutritional symbioses in triatomines: who is playing?Recommended by Natacha Kremer based on reviews by Alejandro Manzano Marín and Olivier DuronNearly 8 million people are suffering from Chagas disease in the Americas. The etiological agent, Trypanosoma cruzi, is mainly transmitted by triatomine bugs, also known as kissing or vampire bugs, which suck blood and transmit the parasite through their feces. Among these triatomine species, Rhodnius prolixus is considered the main vector, and many studies have focused on characterizing its biology, physiology, ecology and evolution. Interestingly, given that Rhodnius species feed almost exclusively on blood, their diet is unbalanced, and the insects can lack nutrients and vitamins that they cannot synthetize themself, such as B-vitamins. In all insects feeding exclusively on blood, symbioses with microbes producing B-vitamins (mainly biotin, riboflavin and folate) have been widely described (see review in Duron and Gottlieb 2020) and are critical for insect development and reproduction. These co-evolved relationships between blood feeders and nutritional symbionts could now be considered to develop new control methods, by targeting the ‘Achille’s heel’ of the symbiotic association (i.e., transfer of nutrient and / or control of nutritional symbiont density). But for this, it is necessary to better characterize the relationships between triatomines and their symbionts. R. prolixus is known to be associated with several symbionts. The extracellular gut symbiont Rhodococcus rhodnii, which reaches high bacterial densities and is almost fixed in R. prolixus populations, appears to be a nutritional symbiont under many blood sources. This symbiont can provide B-vitamins such as biotin (B7), niacin (B3), thiamin (B1), pyridoxin (B6) or riboflavin (B2) and can play an important role in the development and the reproduction of R. prolixus (Pachebat et al. (2013) and see review in Salcedo-Porras et al. (2020)). This symbiont is orally acquired through egg smearing, ensuring the fidelity of transmission of the symbiont from mother to offspring. However, as recently highlighted by Tobias et al. (2020) and Gilliland et al. (2022), other gut microbes could also participate to the provision of B-vitamins, and R. rhodnii could additionally provide metabolites (other than B-vitamins) increasing bug fitness. In the study from Filée et al., the authors focused on Wolbachia, an intracellular, maternally inherited bacterium, known to be a nutritional symbiont in other blood-sucking insects such as bedbugs (Nikoh et al. 2014), and its potential role in vitamin provision in triatomine bugs. After screening 17 different triatomine species from the 3 phylogenetic groups prolixus, pallescens and pictipes, they first show that Wolbachia symbionts are widely distributed in the different Rhodnius species. Contrary to R. rhodnii that were detected in all samples, Wolbachia prevalence was patchy and rarely fixed. The authors then sequenced, assembled, and compared 13 Wolbachia genomes from the infected Rhodnius species. They showed that all Wolbachia are phylogenetically positioned in the supergroup F that contains wCle (the Wolbachia from bedbugs). In addition, 8 Wolbachia strains (out of 12) encode a biotin operon under strong purifying selection, suggesting the preservation of the biological function and the metabolic potential of Wolbachia to supplement biotin in their Rhodnius host. From the study of insect genomes, the authors also evidenced several horizontal transfers of genes from Wolbachia to Rhodnius genomes, which suggests a complex evolutionary interplay between vampire bugs and their intracellular symbiont. This nice piece of work thus provides valuable information to the fields of multiple partners / nutritional symbioses and Wolbachia research. Dual symbioses described in insects feeding on unbalanced diets generally highlight a certain complementarity between symbionts that ensure the whole nutritional complementation. The study presented by Filée et al. leads rather to consider the impact of multiple symbionts with different lifestyles and transmission modes in the provision of a specific nutritional benefit (here, biotin). Because of the low prevalence of Wolbachia in certain species, a “ménage à trois” scenario would rather be replaced by an “open couple”, where the host relationship with new symbiotic partners (more or less stable at the evolutionary timescale) could provide benefits in certain ecological situations. The results also support the potential for Wolbachia to evolve rapidly along a continuum between parasitism and mutualism, by acquiring operons encoding critical pathways of vitamin biosynthesis. References Duron O. and Gottlieb Y. (2020) Convergence of Nutritional Symbioses in Obligate Blood Feeders. Trends in Parasitology 36(10):816-825. https://doi.org/10.1016/j.pt.2020.07.007 Filée J., Agésilas-Lequeux K., Lacquehay L., Bérenger J.-M., Dupont L., Mendonça V., Aristeu da Rosa J. and Harry M. (2023) Wolbachia genomics reveals a potential for a nutrition-based symbiosis in blood-sucking Triatomine bugs. bioRxiv, 2022.09.06.506778, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.09.06.506778 Gilliland C.A. et al. (2022) Using axenic and gnotobiotic insects to examine the role of different microbes on the development and reproduction of the kissing bug Rhodnius prolixus (Hemiptera: Reduviidae). Molecular Ecology. https://doi.org/10.1111/mec.16800 Nikoh et al. (2014) Evolutionary origin of insect–Wolbachia nutritional mutualism. PNAS. 111(28):10257-10262. https://doi.org/10.1073/pnas.1409284111 Pachebat, J.A. et al. (2013). Draft genome sequence of Rhodococcus rhodnii strain LMG5362, a symbiont of Rhodnius prolixus (Hemiptera, Reduviidae, Triatominae), the principle vector of Trypanosoma cruzi. Genome Announc. 1(3):e00329-13. https://doi.org/10.1128/genomea.00329-13 Salcedo-Porras N., et al. (2020). The role of bacterial symbionts in Triatomines: an evolutionary perspective. Microorganisms. 8:1438. https://doi.org/10.3390%2Fmicroorganisms8091438 Tobias N.J., Eberhard F.E., Guarneri A.A. (2020) Enzymatic biosynthesis of B-complex vitamins is supplied by diverse microbiota in the Rhodnius prolixus anterior midgut following Trypanosoma cruzi infection. Computational and Structural Biotechnology Journal. 3395-3401. https://doi.org/10.1016/j.csbj.2020.10.031 | Wolbachia genomics reveals a potential for a nutrition-based symbiosis in blood-sucking Triatomine bugs | Jonathan Filée, Kenny Agésilas-Lequeux, Laurie Lacquehay, Jean Michel Bérenger, Lise Dupont, Vagner Mendonça, João Aristeu da Rosa, Myriam Harry | <p>The nutritional symbiosis promoted by bacteria is a key determinant for adaptation and evolution of many insect lineages. A complex form of nutritional mutualism that arose in blood-sucking insects critically depends on diverse bacterial symbio... | | Genome Evolution, Phylogenetics / Phylogenomics, Species interactions | Natacha Kremer | Alejandro Manzano Marín | 2022-09-13 17:36:46 | |
02 Feb 2023
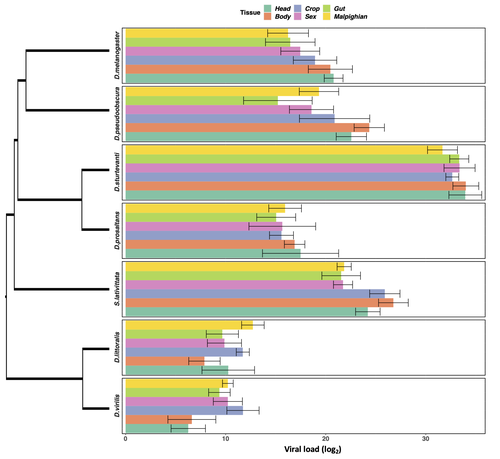
Heterogeneities in infection outcomes across species: sex and tissue differences in virus susceptibilitySusceptibility to infection is not explained by sex or differences in tissue tropism across different species of DrosophilaRecommended by Alison Duncan based on reviews by Greg Hurst and 1 anonymous reviewerUnderstanding factors explaining both intra and interspecific variation in susceptibility to infection by parasites remains a key question in evolutionary biology. Within a species variation in susceptibility is often explained by differences in behaviour affecting exposure to infection and/or resistance affecting the degree by which parasite growth is controlled (Roy & Kirchner, 2000, Behringer et al., 2000). This can vary between the sexes (Kelly et al., 2018) and may be explained by the ability of a parasite to attack different organs or tissues (Brierley et al., 2019). However, what goes on within one species is not always relevant to another, making it unclear when patterns can be scaled up and generalised across species. This is also important to understand when parasites may jump hosts, or identify species that may be susceptible to a host jump (Longdon et al., 2015). Phylogenetic distance between hosts is often an important factor explaining susceptibility to a particular parasite in plant and animal hosts (Gilbert & Webb, 2007, Faria et al., 2013). In two separate experiments, Roberts and Longdon (Roberts & Longdon, 2022) investigated how sex and tissue tropism affected variation in the load of Drosophila C Virus (DCV) across multiple Drosophila species. DCV load has been shown to correlate positively with mortality (Longdon et al., 2015). Overall, they found that load did not vary between the sexes; within a species males and females had similar DCV loads for 31 different species. There was some variation in levels of DCV growth in different tissue types, but these too were consistent across males for 7 species of Drosophila. Instead, in both experiments, host phylogeny or interspecific variation, explained differences in DCV load with some species being more infected than others. This study is neat in that it incorporates and explores simultaneously both intra and interspecific variation in infection-related life-history traits which is not often done (but see (Longdon et al., 2015, Imrie et al., 2021, Longdon et al., 2011, Johnson et al., 2012). Indeed, most studies to date explore either inter-specific differences in susceptibility to a parasite (it can or can’t infect a given species) (Davies & Pedersen, 2008, Pfenning-Butterworth et al., 2021) or intra-specific variability in infection-related traits (infectivity, resistance etc.) due to factors such as sex, genotype and environment (Vale et al., 2008, Lambrechts et al., 2006). This work thus advances on previous studies, while at the same time showing that sex differences in parasite load are not necessarily pervasive. References Behringer DC, Butler MJ, Shields JD (2006) Avoidance of disease by social lobsters. Nature, 441, 421–421. https://doi.org/10.1038/441421a Brierley L, Pedersen AB, Woolhouse MEJ (2019) Tissue tropism and transmission ecology predict virulence of human RNA viruses. PLOS Biology, 17, e3000206. https://doi.org/10.1371/journal.pbio.3000206 Davies TJ, Pedersen AB (2008) Phylogeny and geography predict pathogen community similarity in wild primates and humans. Proceedings of the Royal Society B: Biological Sciences, 275, 1695–1701. https://doi.org/10.1098/rspb.2008.0284 Faria NR, Suchard MA, Rambaut A, Streicker DG, Lemey P (2013) Simultaneously reconstructing viral cross-species transmission history and identifying the underlying constraints. Philosophical Transactions of the Royal Society B: Biological Sciences, 368, 20120196. https://doi.org/10.1098/rstb.2012.0196 Gilbert GS, Webb CO (2007) Phylogenetic signal in plant pathogen–host range. Proceedings of the National Academy of Sciences, 104, 4979–4983. https://doi.org/10.1073/pnas.0607968104 Imrie RM, Roberts KE, Longdon B (2021) Between virus correlations in the outcome of infection across host species: Evidence of virus by host species interactions. Evolution Letters, 5, 472–483. https://doi.org/10.1002/evl3.247 Johnson PTJ, Rohr JR, Hoverman JT, Kellermanns E, Bowerman J, Lunde KB (2012) Living fast and dying of infection: host life history drives interspecific variation in infection and disease risk. Ecology Letters, 15, 235–242. https://doi.org/10.1111/j.1461-0248.2011.01730.x Kelly CD, Stoehr AM, Nunn C, Smyth KN, Prokop ZM (2018) Sexual dimorphism in immunity across animals: a meta-analysis. Ecology Letters, 21, 1885–1894. https://doi.org/10.1111/ele.13164 Lambrechts L, Chavatte J-M, Snounou G, Koella JC (2006) Environmental influence on the genetic basis of mosquito resistance to malaria parasites. Proceedings of the Royal Society B: Biological Sciences, 273, 1501–1506. https://doi.org/10.1098/rspb.2006.3483 Longdon B, Hadfield JD, Day JP, Smith SCL, McGonigle JE, Cogni R, Cao C, Jiggins FM (2015) The Causes and Consequences of Changes in Virulence following Pathogen Host Shifts. PLOS Pathogens, 11, e1004728. https://doi.org/10.1371/journal.ppat.1004728 Longdon B, Hadfield JD, Webster CL, Obbard DJ, Jiggins FM (2011) Host Phylogeny Determines Viral Persistence and Replication in Novel Hosts. PLOS Pathogens, 7, e1002260. https://doi.org/10.1371/journal.ppat.1002260 Pfenning-Butterworth AC, Davies TJ, Cressler CE (2021) Identifying co-phylogenetic hotspots for zoonotic disease. Philosophical Transactions of the Royal Society B: Biological Sciences, 376, 20200363. https://doi.org/10.1098/rstb.2020.0363 Roberts KE, Longdon B (2023) Heterogeneities in infection outcomes across species: examining sex and tissue differences in virus susceptibility. bioRxiv 2022.11.01.514663, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.11.01.514663 Roy BA, Kirchner JW (2000) Evolutionary Dynamics of Pathogen Resistance and Tolerance. Evolution, 54, 51–63. https://doi.org/10.1111/j.0014-3820.2000.tb00007.x Vale PF, Stjernman M, Little TJ (2008) Temperature-dependent costs of parasitism and maintenance of polymorphism under genotype-by-environment interactions. Journal of Evolutionary Biology, 21, 1418–1427. https://doi.org/10.1111/j.1420-9101.2008.01555.x | Heterogeneities in infection outcomes across species: sex and tissue differences in virus susceptibility | Katherine E Roberts, Ben Longdon | <p style="text-align: justify;">Species vary in their susceptibility to pathogens, and this can alter the ability of a pathogen to infect a novel host. However, many factors can generate heterogeneity in infection outcomes, obscuring our ability t... | | Evolutionary Ecology | Alison Duncan | Anonymous, Greg Hurst | 2022-11-03 11:17:42 |




