
Latest recommendations
| Id | Title * | Authors * | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
03 Jan 2025
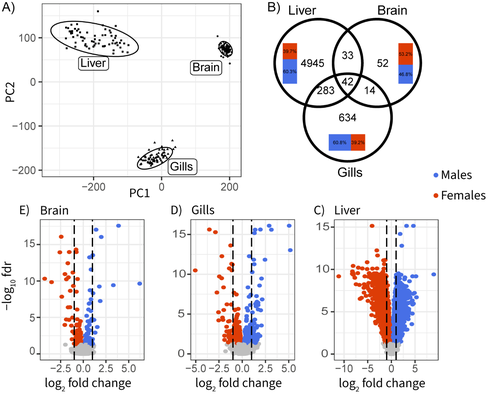
Sex-biased gene expression across tissues reveals unexpected differentiation in the gills of the threespine sticklebackSex-biased gene expression in fish – a milestoneRecommended by Nicolas Galtier based on reviews by Qi Zhou and 2 anonymous reviewers based on reviews by Qi Zhou and 2 anonymous reviewers
There is a heavy body of literature on sex-biased gene expression, which can easily be tricky. One reason is that expression data are multi-dimensional, notoriously noisy, and highly sensitive to experimental conditions. Achieving reproducibility is, therefore, a challenge, especially in non-model organisms. Another reason is that the evolutionary forces shaping gene expression variation are complex, involving processes such as intra- and inter-locus conflicts between sexes, sex-chromosome evolution and degeneration, dosage compensation, cis vs. trans regulation, dominance, linkage, drift, etc... Not surprisingly, the field is rich in discordant studies, and a wide variety of situations have been described. The three-spined stickleback is a good illustration. This widely-studied fish displays conspicuous sexual dimorphisms, both morphological and behavioural. Yet, the existing literature regarding which genes and tissues are involved is remarkably unclear due to the lack of a comprehensive data set. Sylvestre et al. (1) here fix this issue. They sampled 40 wild-caught individuals in each of the two sexes and performed high-throughput sequencing of the transcriptome in three somatic tissues, dissected and preserved in the exact same conditions. This is an impressive effort, well above current standards. Data analysis delivered a series of neat results: gene expression in the liver is particularly sex-biased; the brain, in contrast, is remarkably little sex-differentiated despite the presence of courtship and paternal care in this species; gills show significantly sex-biased gene expression, which had been unnoticed previously despite the importance of this organ in fish ecotoxicology; the relatively young sex-chromosomes, finally, do not seem to experience dosage compensation, and are therefore enriched in sex-biased genes. Some of these results are consistent with previous studies in other fish species (2), here confirmed or demonstrated with a high degree of certainty. Others are new and worth considering in future studies of stickleback ecology and reproduction. We simply need more studies of this sort: well-conducted and clear-cut, recalling the career of its last author. References (1) Florent Sylvestre, Nadia Aubin-Horth, Louis Bernatchez (2024) Sex-biased gene expression across tissues reveals unexpected differentiation in the gills of the threespine stickleback. bioRxiv, ver.2 peer-reviewed and recommended by PCI Evol Biol https://doi.org/10.1101/2024.06.09.597944 (2) Iulia Darolti I, Judith E. Mank (2023). Sex-biased gene expression at single-cell resolution: cause and consequence of sexual dimorphism. Evolution Letters 7(3):148-156. https://doi.org/10.1093/evlett/qrad013
| Sex-biased gene expression across tissues reveals unexpected differentiation in the gills of the threespine stickleback | Florent Sylvestre, Nadia Aubin-Horth, Louis Bernatchez | <p>Sexual dimorphism can evolve through sex-specific regulation of the same gene set. However, sex chromosomes can also facilitate this by directly linking gene expression to sex. Moreover, differences in gene content between heteromorphic sex chr... | | Expression Studies, Genetic conflicts, Genome Evolution, Molecular Evolution, Reproduction and Sex | Nicolas Galtier | 2024-06-11 18:57:06 | ||
18 Dec 2024

Investigating the effects of diurnal and nocturnal pollinators on male and female reproductive success and on floral trait selection in Silene dioicaMore in less: almost everything you wanted to know about sex in flowers is in a single experiment with a single plant speciesRecommended by Juan Arroyo and Violeta Simón-Porcar based on reviews by Luis Gimenez-Benavides, Andrea Cocucci, Giovanni Scopece and 1 anonymous reviewer
Most flowering plants (almost 90% of species) are pollinated by animals (Ollerton et al. 2011). In fact, many plants are completely dependent on pollinator visits for reproductive success, due to the complete inability of selfing if they are self-incompatible or have strong gender differentiation, as in dioecious plants. Others have diminished reproductive output in the absence of pollinators, even being self-compatible, if their flowers present strong herkogamy or dichogamy, making autonomous selfing more difficult. Ultimately, all animal-pollinated plant species rely on pollinators for outcrossing. Depending on the genetic structure of plant populations and the movement patterns of these animals, outcrossing patterns will shape the population genetic variation, which will determine its adaptive fate. Thus, understanding the mechanisms governing the pollination interaction is crucial for unraveling the uncertainties of a huge proportion of biodiversity on Earth. Being mutualistic by definition, the animal side of this interaction is less understood, despite most pollinator groups being likely dependent on it for their persistence and perhaps diversity (Ollerton 2017). The role of pollinators in plant diversification has generated much literature and controversy ever since Darwin and his “abominable mystery” about angiosperm diversification (Friedman 2009). However, the other way around, that of plant`s effect on pollinator diversification, is more debatable. A remarkable example of this effect is the possible case of co-speciation mediated by nursery (brood site) pollination, which also includes antagonistic insect herbivory (Wiens et al. 2015), as in some Silene species and their moth pollinators and herbivores (Hembry and Althoff 2016). The authors of this recommendation benefitted from grants provided by grants PID2021-122715NB-I00 and TED2021-131037B-I00 funded by MCIN/AEI/ 10.13039/501100011033 and by the “European Union NextGeneration EU/PRTR”, and by MSCA-IF-2019-89789. Barbot, E., Dufaÿ, M., Godé, C., & De Cauwer, I. (2024). Exploring the effect of scent emission and exposition to diurnal versus nocturnal pollinators on selection patterns on floral traits. Zenodo. https://doi.org/10.5281/zenodo.11490231 Friedman, W. E. (2009). The meaning of Darwin's “abominable mystery”. American Journal of Botany, 96(1), 5-21. https://doi.org/10.3732/ajb.0800150 Haran, J., Kergoat, G. J., & de Medeiros, B. A. (2023). Most diverse, most neglected: weevils (Coleoptera: Curculionoidea) are ubiquitous specialized brood-site pollinators of tropical flora. Peer Community Journal, 3. https://doi.org/10.24072/pcjournal.279 Hembry, D.H. and Althoff, D.M. (2016), Diversification and coevolution in brood pollination mutualisms: Windows into the role of biotic interactions in generating biological diversity. American Journal of Botany, 103: 1783-1792. https://doi.org/10.3732/ajb.1600056 Kephart, S., Reynolds, R. J., Rutter, M. T., Fenster, C. B., & Dudash, M. R. (2006). Pollination and seed predation by moths on Silene and allied Caryophyllaceae: evaluating a model system to study the evolution of mutualisms. New Phytologist, 169(4), 667-680. https://doi.org/10.1111/j.1469-8137.2005.01619.x Kulbaba, M. W., & Worley, A. C. (2013). Selection on Polemonium brandegeei (Polemoniaceae) flowers under hummingbird pollination: in opposition, parallel, or independent of selection by hawkmoths?. Evolution, 67(8), 2194-2206. https://doi.org/10.1111/evo.12102 Nunes, C. E. P., Maruyama, P. K., Azevedo-Silva, M., & Sazima, M. (2018). Parasitoids turn herbivores into mutualists in a nursery system involving active pollination. Current Biology, 28(6), 980-986. https://doi.org/10.1016/j.cub.2018.02.013 Ollerton, J. (2017). Pollinator diversity: distribution, ecological function, and conservation. Annual review of ecology, evolution, and systematics, 48(1), 353-376. https://doi.org/10.1146/annurev-ecolsys-110316-022919 Ollerton, J., Winfree, R., & Tarrant, S. (2011). How many flowering plants are pollinated by animals?. Oikos, 120(3), 321-326. https://doi.org/10.1111/j.1600-0706.2010.18644.x Prieto-Benitez, S., Yela, J. L., & Gimenez-Benavides, L. (2017). Ten years of progress in the study of Hadena-Caryophyllaceae nursery pollination. A review in light of new Mediterranean data. Flora, 232, 63-72. https://doi.org/10.1016/j.flora.2017.02.004 Raguso, R. A. (2008). Wake up and smell the roses: the ecology and evolution of floral scent. Annual review of ecology, evolution, and systematics, 39(1), 549-569. https://doi.org/10.1146/annurev.ecolsys.38.091206.095601 Simón-Porcar, V. I., Meagher, T. R., & Arroyo, J. (2015). Disassortative mating prevails in style-dimorphic Narcissus papyraceus despite low reciprocity and compatibility of morphs. Evolution, 69(9), 2276-2288. https://doi.org/10.1111/evo.12731 Suetsugu, K. (2023). A novel nursery pollination system between a mycoheterotrophic orchid and mushroom-feeding flies. Ecology, 104(11), e4152. https://doi.org/10.1002/ecy.4152 Wiens, J. J., Lapoint, R. T., & Whiteman, N. K. (2015). Herbivory increases diversification across insect clades. Nature communications, 6(1), 8370. https://doi.org/10.1038/ncomms9370 | Investigating the effects of diurnal and nocturnal pollinators on male and female reproductive success and on floral trait selection in Silene dioica | Barbot Estelle, Dufaÿ Mathilde, Godé Cécile, De Cauwer Isabelle | <p>Plant species with mixed pollination systems are under pollinator-mediated selection by both diurnal and nocturnal pollinator species. This could impact the strength and potentially direction of selection on floral traits, as different pollinat... | | Evolutionary Ecology, Reproduction and Sex | Juan Arroyo | 2024-06-05 15:52:46 | ||
18 Nov 2024

Faster model-based estimation of ancestry proportionsfastmixture generates fast and accurate estimates of global ancestry proportions and ancestral allele frequenciesRecommended by Matteo Fumagalli based on reviews by Oscar Lao Grueso and 2 anonymous reviewers
The estimation of ancestry proportions in individuals is an important analysis in both evolutionary biology and medical genetics. However, popular tools like ADMIXTURE (Alexander et al. 2009) and STRUCTURE (Pritchard et al. 2000) do not scale well with the large amount of data currently available. Recent alternative methods, such as SCOPE (Chiu et al. 2022), favour scalability over accuracy. In this study, Santander and coworkers introduce a new software, called fastmixture, which estimates ancestry proportions and ancestral allele frequencies using novel implementations for initialisation and convergence of its model-based algorithm (Santander et al. 2024). In simulated datasets, fastmixture displays desirable properties of speed and accuracy, with its performance surpassing commonly used software (Alexander et al. 2009, Pritchard et al. 2000, Chiu et al. 2022, Mantes et al. 2023). fastmixture is almost 30 times faster than ADMIXTURE under a complex model with five ancestral populations, while retaining similar accuracy levels. When applied to data from the 1000 Genomes Project (1000 Genomes Project Consortium 2025), fastmixture recapitulated expected levels of global ancestry. The new software is freely available on GitHub with an accessible documentation. fastmixture accepts input files in PLINK format. It remains an open question whether extensive parameter tuning could increase the scalability and accuracy of established methods. A comprehensive assessment of fastmixture over a wide range of data processing options (Hemstrom et al. 2024) is also missing. Finally, whether model-based approaches are fully scalable to ever increasing biobank datasets is still under debate. Nevertheless, the superior computational performance of fastmixture is evident and it is likely that this new software will soon replace existing popular tools to estimate global ancestry proportions. References Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655-1664. https://doi.org/10.1101/gr.094052.109 Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945-959. https://doi.org/10.1093/genetics/155.2.945 Chiu AM, Molloy EK, Tan Z, Talwalkar A, Sankararaman S. Inferring population structure in biobank-scale genomic data. Am J Hum Genet. 2022;109(4):727-737. https://doi.org/10.1016/j.ajhg.2022.02.015 Santander CG, Refoyo Martinez A, Meisner J. Faster model-based estimation of ancestry proportions. bioRxiv 2024; ver.2 peer-reviewed and recommended by PCI Evol Biol. https://doi.org/10.1101/2024.07.08.602454 Mantes AD, Montserrat DM, Bustamante CD, Giró-I-Nieto X, Ioannidis AG. Neural ADMIXTURE for rapid genomic clustering. Nat Comput Sci. 2023;3(7):621-629. https://doi.org/10.1038/s43588-023-00482-7 1000 Genomes Project Consortium, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68-74. https://doi.org/10.1038/nature15393 Hemstrom W, Grummer JA, Luikart G, Christie MR. Next-generation data filtering in the genomics era. Nat Rev Genet. 2024;25(11):750-767. https://doi.org/10.1038/s41576-024-00738-6 | Faster model-based estimation of ancestry proportions | Cindy G. Santander, Alba Refoyo Martinez, Jonas Meisner | <p>Ancestry estimation from genotype data in unrelated individuals has become an essential tool in population and medical genetics to understand demographic population histories and to model or correct for population structure. The ADMIXTURE softw... | | Bioinformatics & Computational Biology, Human Evolution, Population Genetics / Genomics | Matteo Fumagalli | 2024-07-14 11:48:39 | ||
18 Nov 2024
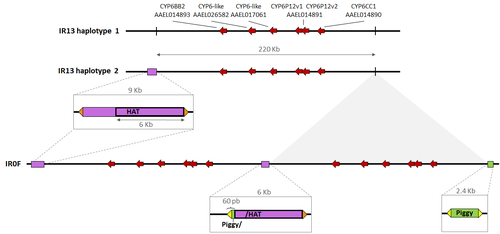
A genomic duplication spanning multiple P450s contributes to insecticide resistance in the dengue mosquito Aedes aegyptiA duplication driving metabolic insecticide resistance in Aedes aegyptiRecommended by Diego A. Hartasánchez based on reviews by Diego Ayala and 1 anonymous reviewer
Insecticide resistance in mosquitoes represents a notable challenge to public health efforts aimed at controlling vector-borne diseases. Among mosquito species, Aedes aegypti is particularly significant due to its extensive geographic spread and its ability to transmit arboviruses causing diseases such as dengue, yellow fever, Zika, and chikungunya (Brown et al., 2014). Insecticide resistance typically develops through two main mechanisms: target-site mutations, which affect the insecticide's interaction with its target, and metabolic resistance, in which insecticide detoxification is enhanced in mosquitoes. While target-site mutations are well characterized, the mechanisms underlying metabolic resistance are understudied. The study by Bacot and colleagues (2024) contributes to our understanding of the genetic and evolutionary mechanisms driving insecticide resistance, focusing on a case of metabolic resistance in Aedes aegypti from French Guiana. Following the recent identification of a copy number variant region on chromosome 1, potentially linked to overexpression of detoxification enzymes (Cattel et al., 2020), this study explores the region’s genomic architecture, its likely origin and provides compelling evidence for its role in insecticide resistance. Through RNA sequencing and whole-genome pool sequencing, the authors reveal that this 220 kilobase duplication increases the expression level of several clustered P450 genes. Cytochrome P450s are known to play a role in breaking down pyrethroids like deltamethrin, a commonly used insecticide. The role of P450 enzymes in detoxification was demonstrated by treating mosquitoes with piperonyl butoxide, a P450 enzyme inhibitor, and observing reduction in deltamethrin resistance, further confirmed by RNA interference experiments. Despite the clear advantages of this genomic duplication in conferring resistance, the study also uncovers a fitness cost associated with carrying the duplication. Through experimental evolution, the authors find that mosquitoes with the duplication experience reduced fitness in the absence of insecticide pressure. Given the regions structural complexity, the authors could not completely disassociate the effect of the duplicated region and that of a target-site mutation. However, they developed an assay that can accurately track the presence of this resistance allele in mosquito populations. Altogether, the study by Bacot et al. (2024) highlights the challenges of characterizing the phenotypic effect of copy number variant regions in complex genomes, such as that of Aedes aegypti. It emphasizes the need for further studies on the origin and spread of this duplication to better understand how similar resistance mechanisms might evolve and disseminate. Overall, the completeness and coherence of the narrative, the detailed and thorough analysis, and the insightful discussion, make this work not only a significant contribution to the field of insecticide resistance but an interesting read for the general evolutionary biology community. References Brown, J. E., Evans, B. R., Zheng, W., Obas, V., Barrera-Martinez, L., Egizi, A., Zhao, H., Caccone, A., & Powell, J. R. (2014). Human impacts have shaped historical and recent evolution in Aedes aegypti, the dengue and yellow fever mosquito. Evolution, 68(2), 514–525. https://doi.org/10.1111/evo.12281 Cattel, J., Faucon, F., Le Péron, B., Sherpa, S., Monchal, M., Grillet, L., Gaude, T., Laporte, F., Dusfour, I., Reynaud, S., & David, J. P. (2019). Combining genetic crosses and pool targeted DNA-seq for untangling genomic variations associated with resistance to multiple insecticides in the mosquito Aedes aegypti. Evolutionary applications, 13(2), 303–317. https://doi.org/10.1111/eva.12867 Tiphaine Bacot, Chloe Haberkorn, Joseph Guilliet, Julien Cattel, Mary Kefi, Louis Nadalin, Jonathan Filee, Frederic Boyer, Thierry Gaude, Frederic Laporte, Jordan Tutagata, John Vontas, Isabelle Dusfour, Jean-Marc Bonneville, Jean-Philippe David (2024) A genomic duplication spanning multiple P450s contributes to insecticide resistance in the dengue mosquito Aedes aegypti. bioRxiv, ver.5 peer-reviewed and recommended by PCI Evol Biol https://doi.org/10.1101/2024.04.03.587871 | A genomic duplication spanning multiple P450s contributes to insecticide resistance in the dengue mosquito *Aedes aegypti* | Tiphaine Bacot, Chloe Haberkorn, Joseph Guilliet, Julien Cattel, Mary Kefi, Louis Nadalin, Jonathan Filee, Frederic Boyer, Thierry Gaude, Frederic Laporte, Jordan Tutagata, John Vontas, Isabelle Dusfour, Jean-Marc Bonneville, Jean-Philippe David | <p>Resistance of mosquitoes to insecticides is one example of rapid adaptation to anthropogenic selection pressures having a strong impact on human health and activities. Target-site modification and increased insecticide detoxification are the tw... | | Adaptation, Evolutionary Applications, Expression Studies, Genotype-Phenotype | Diego A. Hartasánchez | 2024-04-10 11:36:06 | ||
24 Oct 2024

Cryptic species and hybridisation in corals: challenges and opportunities for conservation and restorationHow common cryptic coral diversity can blur biodiversity metrics and challenge managementRecommended by Eric Pante based on reviews by 2 anonymous reviewers
Biological conservation aims at protecting the genetic diversity generated by evolutionary processes over the course of life's history on earth (Allendorf and Luikart 2007), and to be effective, it requires that its fundamental units (among which populations and species) be delimited as precisely as possible. This exercise is particularly important for corals because hybridisation and introgression have played a fundamental role in shaping their contemporary diversity (eg Veron 1995). In their review paper, Riginos et al (2024) show that 68% of nominal taxa investigated for genomic population structure bear the molecular signature of partial reproductive isolation, and can be considered as cryptic genetic groups. Another review study (published a day before the preprint of Riginos et al), converges in the finding that cryptic diversity is rampant in nominal coral species (Grupstra et al 2024). This result has strong bearing on the study of coral biology; as Riginos et al state, "any coral investigation that does not genotype the corals under study risks treating a heterogeneous mix of partially reproductively isolated taxa as a single species." The stakes are therefore high, given the ecological importance of corals and the ecosystem services they provide. While Grupstra et al (2024) discusses the impact of cryptic coral diversity in the context of functional differences in thermal adaptation and the processes that lead to cryptic lineages, Riginos et al (2024) provide a quantitative review of cryptic lineages within nominal species, providing reproducible criteria for delineation, details the importance of detecting hybrids, summarises how biodiversity metrics and conservation efforts can be biased by unrecognised cryptic lineages, offer recommendations on how to recognise and deal with cryptic diversity, and discuss how corals can be regarded as highly valuable model systems to study adaptation and speciation. The study of Riginos et al (2024) is therefore a must-read to all coral biologists, especially those involved in biological conservation. References Fred W. Allendorf and Gordon Luikart (2007) Conservation and Genetics of Populations. Blackwell Publishing, Malden MA, USA. Carsten G. B. Grupstra, Matías Gómez-Corrales, James E. Fifer, Hannah E. Aichelman, Kirstin S. Meyer-Kaiser, Carlos Prada, Sarah W. Davies (2024) Integrating cryptic diversity into coral evolution, symbiosis and conservation. Nat Ecol Evol 8, 622–636 (2024). https://doi.org/10.1038/s41559-023-02319-y Cynthia Riginos, Iva Popovic, Zoe Meziere, Vhon Garcia, Ilha Byrne, Samantha Howitt, Hisatake Ishida, Kevin Bairos-Novak, Adriana Humanes, Hugo Scharfenstein, Thomas Richards, Ethan Briggs, Vanessa Clark, Chuan Lei, Mariam Khan, Katharine Prata (2024) Cryptic species and hybridisation in corals: challenges and opportunities for conservation and restoration. EcoEvoRxiv, ver.2 peer-reviewed and recommended by PCI Evol Biol https://doi.org/10.32942/X2502X J. E. N. Veron (1995) Corals in Space and Time: The Biogeography and Evolution of the Scleractinia. Cornell University Press | Cryptic species and hybridisation in corals: challenges and opportunities for conservation and restoration | Cynthia Riginos, Iva Popovic, Zoe Meziere, Vhon Garcia, Ilha Byrne, Samantha Howitt, Hisatake Ishida, Kevin Bairos-Novak, Adriana Humanes, Hugo Scharfenstein, Thomas Richards, Ethan Briggs, Vanessa Clark, Chuan Lei, Mariam Khan, Katharine Prata | <p style="text-align: justify;">The conservation and management of coral reef ecosystems will benefit from accurate assessments of reef-building coral species diversity. However, the true diversity of corals may be obfuscated by cryptic yet geneti... | | Adaptation, Evolutionary Applications, Hybridization / Introgression, Population Genetics / Genomics, Speciation | Eric Pante | 2024-02-15 10:29:46 | ||
26 Sep 2024
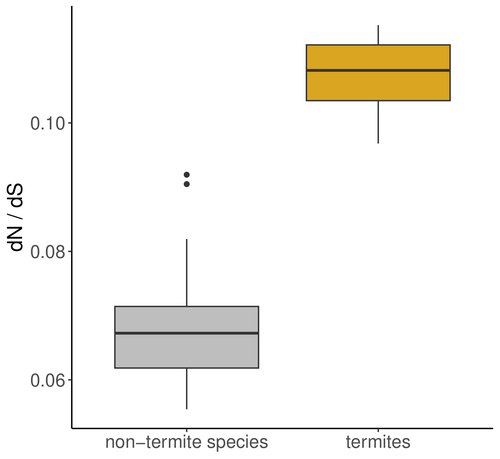
The impact of social complexity on the efficacy of natural selection in termitesEvolutionary trajectories of social transitions: Higher social complexity is associated with lower effective population size and reduced efficacy of selection in termitesRecommended by Trine Bilde based on reviews by 2 anonymous reviewers
A comprehensive study by Roux et al 2024 investigates the impact of eusociality on the efficacy of natural selection in termites, with and additional focus on whether higher levels of social complexity are associated with lower effective population size (Ne) and relaxed purifying selection. Eusociality is characterized by a division of reproductive labor, cooperative care of offspring, and overlapping generations, and has evolved independently across various animal taxa with the most complex social systems found in Hymenoptera (bees, wasps, ants) and termites. Because reproduction is limited to a few individuals, this leads to a reduced effective population size (Ne), which impacts genome evolution. Smaller Ne increases the influence of genetic drift, weakening the efficiency of natural selection and allowing the accumulation of weakly deleterious mutations. This phenomenon, known as the "drift barrier," alters the mutation-selection balance in eusocial organisms. Studies in a range of social arthropods including ants, termites, crustaceans and spiders have shown an elevated ratio of nonsynonymous to synonymous substitutions (dN/dS), indicating relaxed purifying selection due to small Ne. In termite species with complex social structures, such as those with large colonies and high caste specialization, there are reports of higher dN/dS ratios compared to simpler social species. This suggests that higher social complexity, reflected in traits like nesting strategies and developmental pathways, further reduces Ne and the effectiveness of natural selection. The authors address these hypotheses by exploring the genomic impact of eusociality in termites (Isoptera) taking two approaches: First, they analyze transcriptome data from 66 Blattodea species and calculate the ratio of non-synonymous to synonymous mutations (dN/dS) as an indicator of natural selection efficiency and effective population size. They analyses reveal an increased dN/dS ratio in termites compared to other Blattodea species, reinforcing the notion that convergent evolution toward eusociality significantly reduces effective population size and weakens natural selection efficiency across the genome. Additionally, a comparison of 68 termite transcriptomes shows that this effect is more pronounced in species with higher social complexity. This is exciting as it advances our understanding of how increasing complexity in social organization decreases Ne and the efficiency of natural selection. The study substantiates the notion that social transitions follow evolutionary trajectories where lower and Ne and increasing drift have negative consequences for genome evolution (Ma et al 2024). References Camille Roux, Alice Ha, Arthur Weyna, Morgan Lode, Jonathan Romiguier (2024) The impact of social complexity on the efficacy of natural selection in termites. bioRxiv, ver.2 peer-reviewed and recommended by PCI Evol Biol. https://doi.org/10.1101/2024.04.26.591327 Jilong Ma, Jesper Bechsgaard, Anne Aagaard, Palle Villesen, Trine Bilde, Mikkel Heide Schierup (2024) Sociality in spiders is an evolutionary dead-end. | The impact of social complexity on the efficacy of natural selection in termites | Camille Roux, Alice Ha, Arthur Weyna, Morgan Lode, Jonathan Romiguier | <p style="text-align: justify;">In eusocial species, reproduction is monopolized by a few reproductive individuals. From the perspective of population genetics, this implies that the effective population size (Ne<em>)</em> of these organisms is li... | | Molecular Evolution | Trine Bilde | 2024-04-30 12:10:20 | ||
24 Sep 2024
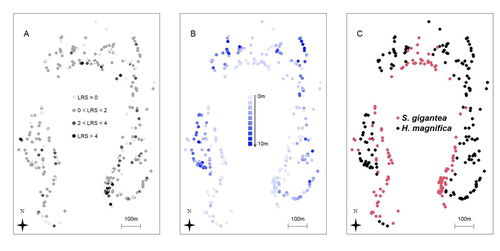
Spatial autocorrelation and host anemone species drive variation in local components of fitness in a wild clownfish populationIs our best measure of fitness correlated with environment? A study in an orange clownfish population.Recommended by Pierre de Villemereuil based on reviews by Stefan Vriend and 2 anonymous reviewers
Getting a clear definition of fitness for a particular evolutionary biology question is a complex challenge, fraught with pitfalls and misconceptions (Orr, 2009; Walsh & Lynch, 2018). In longitudinal surveys of wild populations, lifetime reproductive success (LRS) is generally considered the best measure of individual fitness (Bonnet, 2022). However, it is important to bear in mind that LRS is only a (noisy) measure of the realised success, relying on a substantial amount of assumptions (e.g. with regard to generation overlap, Walsh & Lynch, 2018), not a direct measure of fitness. In a study on the clownfish, Marrot et al. (2024) studied the spatial and ecological drivers of lifetime reproductive success. To do so, they analysed a 10-year long survey on over 300 anemones harbouring clownfishes, and used a genetics-based pedigree to infer the LRS of each individual. Using a characterisation of the micro-habitat provided by each anemone, they used the anemone species, density and depth as ecological drivers and spatial-autocorrelated models to study more general (and undefined) spatial drivers. The authors found that LRS was influenced by a significant amount by the spatial structure of the population, and, to some extent, by the anemone species harbouring the clownfish individuals. Together, they explain a substantial proportion of the individual variation in LRS. While the actual determinants of spatial variation of LRS in this (and other) species remain understood, this study highlights an important aspect of measuring fitness in wild populations using LRS: it is particularly noisy and subject to environmental variation. This certainly does not mean that LRS is a bad proxy for fitness, it is still among the best measure of it we can have access to. However, it highlights how carefully we should thread when analysing it. Especially, spatial auto-correlation of LRS, combined with population structure within a population, would lead to genotype-environment correlation for fitness, which is likely to bias predictions of response to natural selection and would be extremely difficult to estimate (Falconer & Mackay, 1996). References Pascal Marrot, Cécile Fauvelot, Michael L. Berumen, Maya Srinivasan, Geoffrey P. Jones, Serge Planes, and Benoit Pujol (2024) Spatial autocorrelation and host anemone species drive variation in local components of fitness in a wild clownfish population. Zenodo, ver.3 peer-reviewed and recommended by PCI Evol Biol https://doi.org/10.5281/zenodo.13806778 Bonnet, T., Morrissey, M. B., de Villemereuil, P., Alberts, S. C., Arcese, P., Bailey, L. D., Boutin, S., Brekke, P., Brent, L. J. N., Camenisch, G., Charmantier, A., Clutton-Brock, T. H., Cockburn, A., Coltman, D. W., Courtiol, A., Davidian, E., Evans, S. R., Ewen, J. G., Festa-Bianchet, M., … Kruuk, L. E. B. (2022). Genetic variance in fitness indicates rapid contemporary adaptive evolution in wild animals. Science, 376(6596), 1012–1016. https://doi.org/10.1126/science.abk0853 Falconer, D. S. and Mackay, T. F. C. (1996). Introduction to quantitative genetics (4th ed.). Benjamin Cummings. Orr, H. A. (2009). Fitness and its role in evolutionary genetics. Nature Reviews Genetics, 10(8), 531–539. https://doi.org/10.1038/nrg2603 Walsh, B. and Lynch, M. (2018). Evolution and selection of quantitative traits. Oxford University Press. | Spatial autocorrelation and host anemone species drive variation in local components of fitness in a wild clownfish population | Pascal Marrot, Cécile Fauvelot, Michael L. Berumen, Maya Srinivasan, Geoffrey P. Jones, Serge Planes, and Benoit Pujol | <p style="text-align: justify;">The susceptibility of species to habitat changes depends on which ecological drivers shape individual fitness components. To date, only a few studies have quantified fitness components such as the Lifetime Reproduct... | | Adaptation, Evolutionary Ecology, Quantitative Genetics | Pierre de Villemereuil | 2023-07-31 11:42:58 | ||
26 Aug 2024

Reproductive modes in populations of late-acting self-incompatible and self-compatible polyploid Ludwigia grandiflora subsp. hexapetala in western EuropeMixed reproduction modes explain a high genetic diversity in the invasive plant Ludwigia grandiflora subsp. hexapetala in western EuropeRecommended by Ines Alvarez based on reviews by Rubén Torices and 2 anonymous reviewers
The introduction of Ludwigia species as ornamental plants in both North America and Europe dates back almost two centuries, during which time they expanded as naturalized and later invasive species in these territories (Dandelot et al. 2005, Okada et al. 2009). Repeated deliberate or non-deliberate introductions over time of this species complex that can hybridize has given rise to an evolutionarily complex scenario, which is compounded by the difficulty in delimiting some of these species and by the diversity of their modes of reproduction. Dandelot (2004) and Dandelot et al. (2005) determined the presence of two Ludwigia taxa in France, L. peploides subsp. montevidensis (Spreng.) P.H.Raven (here after Lpm), and L. grandiflora subsp. hexapetala (Hook. & Arn.) G.L.Nesom & Kartesz (here after Lgh), based on their cytotypes (2n = 16, and 2n = 80, respectively) and without evidence of hybridization between them. Furthermore, despite a predominantly vegetative reproduction observed for both species, they differed in their breeding systems. While Lpm is self-compatible and produce a high number of viable seeds in all populations, Lgh is self-incompatible and its populations may drastically differ in seed viability (Dandelot 2004). Several years later, Portillo-Lemus et al. (2021) determined that the differences in seed production between some populations of Lgh are due to the existence of a heteromorphic reproductive system in this taxon, involving a self-incompatible morph (long-style morph; hereafter L-morph), and a self-compatible morph (short-style morph: hereafter S-morph). Moreover, Portillo-Lemus et al. (2022) observed that self-pollen in the L-morph flowers stop growing lately (i.e., in the ovarian area) without fertilizing the ovules, concluding that a late-acting self-incompatible system (hereafter, LSI) is present in this morph. At this point, it is relevant to understand the possible interactions between populations of different morphs in Lgh, and the implications that they may have on their expansive success in non-native areas in order to develop more effective management plans. To achieve this goal, Stoeckel et al. (2024) analyzed the population genetics in 53 Lgh populations in western Europe (without finding any Lpm population in the sampling area), 40 of which exclusively presented the L-morph and 13 the S-morph. This fact offered the opportunity to compare and interpret the differences between populations of different morphs in Lgh. Other previous works on genetic diversity of Lgh in peripatric or non-native areas pointed to a high clonality and an extremely low genetic diversity (Okada et al. 2009, Armitage et al. 2013), concluding in a monoclonal or few ancestral original clones for these invasive populations. However, the investigations of Stoeckel et al. (2024) found a high genetic diversity in all populations of Lgh studied despite their predominant clonal reproduction. Interestingly, they found that sexual reproduction is also present, not only in the S-morph by selfing, but also in the L-morph, although limited and preferably by allogamy. They discuss the advantages and drawbacks of the different modes of reproduction observed in Lgh populations, the interactions among them, and the implications that both, the scarcely documented LSI (Gibbs 2014) and selfing, have in the reproductive success and in the maintenance of the high genetic diversity observed in Lgh in western Europe. The contrasting results with the previous ones (Okada et al. 2009, Armitage et al. 2013) stress the relevance of using appropriate markers and analyses to assess the genetic diversity in autoployploid species, as well as the necessity of knowing the modes of reproduction in the populations studied for an optimal interpretation of the genetic metrics. The approach of the study by Stoeckel et al. (2024) had the challenge of having found suitable markers to deal with a taxon of complex origin such as Lgh, whose genome is compound by a set of autotetraploid chromosomes shared with Lpm and traces of ancient hybridizations of other diploid lineages (Barloy et al. 2024). Using RAD-Seq, Stoeckel et al. (2024) generated an original set of 36 polymorphic SNPs shared between Lgh and Lpm ensuring that these SNPs belong to the tetraploid part of the Lgh genome derived from Lpm. Another interesting contribution of this work is the exhaustive analysis of several genetic descriptors (indexes) and the interpretative guide they provide for each of them in relation to the different modes of reproduction of the study system. Finally, they propose a pair of very useful synthetic indices (i.e., clonality index and selfing index), since they allow to classify populations according to their levels of clonality and selfing. Stoeckel et al. (2024) conclude the relevance that selfing and LSI populations, and the hybridization between them may have on the expansion and success of invasive plant species, and the necessity to know the modes of reproduction of these populations jointly with their genetic diversity in order to develop appropriate management plans. This study raises new questions such as the modes of reproduction and genetic diversity and structure have other Lgh populations, both invasive and native, and the dynamics of these populations under different future scenarios. References Armitage, J. D., Könyves, K., Bailey, J. P., David, J. C., & Culham, A. (2013). A molecular, morphological and cytological investigation of the identity of non-native Ludwigia (Onagraceae) populations in Britain. New Journal of Botany, 3(2), 88–95. https://doi.org/10.1179/2042349713Y.0000000023 Barloy, D., Lemus, L. P.-, Krueger-Hadfield, S. A., Huteau, V., & Coriton, O. (2024). Genomic relationships among diploid and polyploid species of the genus Ludwigia L. section Jussiaea using a combination of cytogenetic, morphological, and crossing investigations. ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.01.02.522458 Dandelot, S. (2004). Les Ludwigia spp. invasives du Sud de la France: Historique, Biosystématique, Biologie et Ecologie [PhD thesis, Aix-Marseille 3]. https://www.theses.fr/2004AIX30052 Dandelot, S., Verlaque, R., Dutartre, A., & Cazaubon, A. (2005). Ecological, dynamic and taxonomic problems due to Ludwigia (Onagraceae) in France. Hydrobiologia, 551(1), 131–136. https://doi.org/10.1007/s10750-005-4455-0 Gibbs, P. E. (2014). Late-acting self-incompatibility – the pariah breeding system in flowering plants. New Phytologist, 203(3), 717–734. https://doi.org/10.1111/nph.12874 Okada, M., Grewell, B. J., & Jasieniuk, M. (2009). Clonal spread of invasive Ludwigia hexapetala and L. grandiflora in freshwater wetlands of California. Aquatic Botany, 91(3), 123–129. https://doi.org/10.1016/j.aquabot.2009.03.006 Portillo Lemus, L. O., Bozec, M., Harang, M., Coudreuse, J., Haury, J., Stoeckel, S., & Barloy, D. (2021). Self-incompatibility limits sexual reproduction rather than environmental conditions in an invasive water primrose. Plant-Environment Interactions, 2(2), 74–86. https://doi.org/10.1002/pei3.10042 Portillo Lemus, L. O., Harang, M., Bozec, M., Haury, J., Stoeckel, S., & Barloy, D. (2022). Late-acting self-incompatible system, preferential allogamy and delayed selfing in the heteromorphic invasive populations of Ludwigia grandiflora subsp. hexapetala. Peer Community Journal, 2. https://doi.org/10.24072/pcjournal.108 Stoeckel, S., Becheler, R., Portillo-Lemus, L., Harang, M., Besnard, A.-L., Lassalle, G., Causse-Védrines, R., Michon-Coudouel, S., Park D. J., Pope, B. J., Petit, E. J. & Barloy, D. (2024) Reproductive modes in populations of late-acting self-incompatible and self-compatible polyploid Ludwigia grandiflora subsp. hexapetala in western Europe. biorxiv, ver.4 peer-reviewed and recommended by PCI Evol Biol https://doi.org/10.1101/2024.03.21.586104 | Reproductive modes in populations of late-acting self-incompatible and self-compatible polyploid *Ludwigia grandiflora* subsp. hexapetala in western Europe | Solenn Stoeckel, Ronan Becheler, Luis Portillo-Lemus, Marilyne Harang, Anne-Laure Besnard, Gilles Lassalle, Romain Causse-Védrines, Sophie Michon-Coudouel, Daniel J. Park, Bernard J. Pope, Eric J. Petit, Dominique Barloy | <p>Reproductive mode, i.e., the proportion of individuals produced by clonality, selfing and outcrossing in populations, determines how hereditary material is transmitted through generations. It shapes genetic diversity and its structure over time... | | Evolutionary Applications, Evolutionary Ecology, Population Genetics / Genomics, Reproduction and Sex | Ines Alvarez | 2024-03-25 10:33:17 | ||
25 Jun 2024
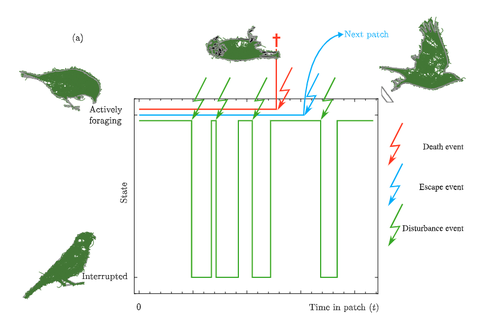
Taking fear back into the Marginal Value Theorem: the risk-MVT and optimal boldnessApplying the marginal value theorem when risk affects foraging behaviorRecommended by Stephen Proulx based on reviews by Taom Sakal and 1 anonymous reviewer
Foraging has been long been studied from an economic perspective, where the costs and benefits of foraging decisions are measured in terms of a single currency of energy which is then taken as a proxy for fitness. A mainstay foraging theory is Charnov’s Marginal Value Theorem (Charnov, 1976), or MVT, which includes a graphical interpretation and has been applied to an enormous range topics in behavioral ecology (Menezes , 2022). Empirical studies often find that animals deviate from MVT, sometimes in that they predictably stay longer than the optimal time. One explanation for this comes from state based models of behavior (Nonacs 2001) Now Calcgano and colleagues (2024) set out to extend and unify foraging models that include various aspects of risk to the foragers, and propose using a risk MVT, or rMVT. They consider three types of risk that foragers face, disturbance, escape, and death. Disturbance represents scenarios where the forager is either physically interrupted in their foraging, or stops foraging temporarily because of the presence of a predator (i.e. a fear response). Such a disturbance can be thought of as altering the gain function for resources acquired while foraging in the patch, allowing the rMVT to be applied in a familiar way with only a reinterpretation of the gain function. In the escape scenarios, foragers are forced to leave a patch because of predator behavior, and therefore artificially decrease their foraging time as compared with their desired foraging time. Now, optimization can be calculated based on this expected time foraging, which means that in effect the forager compensates for the reduced time in the patch by modifying their view of how long they will actually forage. Finally they consider scenarios where risk may result in death, and further divide this into two cases, one where foraging returns are instantaneously converted to fitness, and another where they are only converted in between foraging bouts. This represents an important case to consider, because the total number of foraging trips now depends on the rate of predator attack. In these scenarios, the boldness of the forager is decreased and they become more risk-averse. The authors find that under the disturbance and escape scenarios, patch residence time can actually go up with risk. This is in effect because they are depleting the patch less per unit time, because a larger fraction of time is taken up with avoiding predators. In terms of field applications, this may differ from what is typically considered as risk, since harassment by conspecifics has the same disturbance effect as predator avoidance behaviors. Most experiments on foraging are done in the absence of risk or signals of risk, i.e. in laboratory or otherwise controlled environments. The rMVT predictions deviate from non-risk scenarios in complex ways, in that the patch residence time may increase or decrease under risk. It is also important to note that foragers have evolved their foraging strategies in response to the risk profiles that they have historically experienced, and therefore experiments lacking risk may still show that foragers alter their behavior from the MVT predictions in a way that reflects historical levels of risk. References Calcagno, V., Grognard, F., Hamelin, F.M. and Mailleret, L. (2024). Taking fear back into the Marginal Value Theorem: the risk-MVT and optimal boldness. bioRxiv, 2023.10.31.564970, ver. 3 peer-reviewed and recommended by PCI Evolutionary Biology. https://doi.org/10.1101/2023.10.31.564970 Charnov E. (1976). Optimal foraging the marginal value theorem. Theor Popul Biol. 9, 129–136. Menezes, JFS (2022).The marginal value theorem as a special case of the ideal free distribution. Ecological Modelling 468:109933. https://doi.org/10.1016/j.ecolmodel.2022.109933 Nonacs, P. 2001. State dependent behavior and the Marginal Value Theorem. Behavioral Ecology 12(1) 71–83. https://doi.org/10.1093/oxfordjournals.beheco.a000381 | Taking fear back into the Marginal Value Theorem: the risk-MVT and optimal boldness | Vincent Calcagno, Frederic Grognard, Frederic M Hamelin, Ludovic Mailleret | <p>Foragers exploiting heterogeneous habitats must make strategic movement decisions in order to maximize fitness. Foraging theory has produced very general formalizations of the optimal patch-leaving decisions rational individuals should make. On... | | Adaptation, Behavior & Social Evolution, Evolutionary Ecology, Evolutionary Theory, Life History | Stephen Proulx | 2023-11-03 13:25:16 | ||
31 May 2024
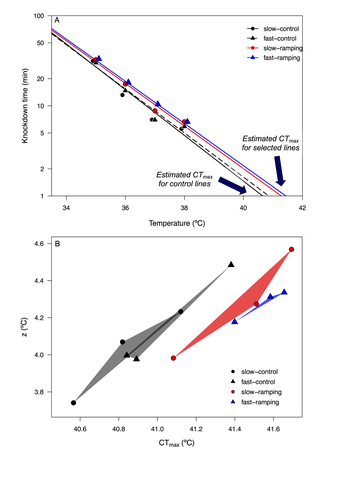
Cross-tolerance evolution is driven by selection of heat tolerance in Drosophila subobscuraEvolution of cross-tolerance: a mechanism to cope with climate change?Recommended by Pedro Simões based on reviews by Marina Stamenkovic-Radak and 1 anonymous reviewer
Understanding how populations evolve under thermal stress and how this process shapes the response of other stress responses is an important research topic in the context of thermal adaptation and climate change. In a thermal experimental evolution study in Drosophila subobscura, Castañeda (2024) addressed the correlated responses to selection for increasing knockdown temperature in different resistance traits, either directly related to thermal stress (e.g. knockdown time at different temperatures and CTmax) or not (e.g. desiccation and starvation resistance). The author found that the evolution of higher knockdown temperature did in fact lead to correlated responses in other stress traits. While such correlations might be expected for the thermal stress traits measured (knockdown time and CTmax), it was perhaps less expectable for desiccation and starvation resistance. However, the general occurrence of correlated evolutionary responses between stressors has been previously described, namely in Drosophila (e.g. see Bubliy and Loeschcke 2005), pointing to a possible genetic link between distinct (thermal) stress traits. There are however some features that make the findings of this study rather appealing. First, the evidence that the correlated stress responses depend on the intensity of thermal selection (i.e. the warming rate) and on the sex of the organisms. Second, correlated patterns of both desiccation and starvation resistance highlight the possibility of the evolution of a cross-tolerance response, which might positively impact on population ability to evolve under sustained stressful environments (Rodgers and Gomez Izasa 2023). However, it is important to point out that the correlated patterns between these two resistance traits (desiccation and starvation) were not exactly consistent. In fact, the negative correlated response observed for female starvation resistance is thought provoking and argues again a general scenario of cross-tolerance. While these findings are a step forward for a more multifaceted understanding of thermal adaptation in the context of stressful environments, they also highlight the need for further studies of thermal adaptation namely 1) addressing the underlying physiological and genomic mechanisms that link male and female heat tolerance and the response to other stress resistance traits (namely starvation resistance); 2) testing the extent to which cross-resistance patterns can be generalized to different thermal selection contexts and populations. In addition, this study also opens new questions considering the scope of correlated evolution to other stress traits, that might be relevant in diverse ecological scenarios. For instance, does selection towards higher heat resistance lead to correlated evolution of cold resistance? And under which circumstances (e.g. different heat selection intensities)? In fact, the occurrence of a positive (or negative) correlation cold and heat stress responses is a topic of high interest, with relevant ecological implications particularly considering the increased thermal fluctuations in natural environments because of climate warming. Cross-tolerance between cold and heat stress responses has been described (Singh 2022, Rodgers and Gomez Izasa 2023). On the other hand, negative correlations (i.e. trade-offs) between these stress traits (Stazione et al. 2020; Schou et al 2022) can impact negatively on populations’ ability to withstand thermal variability. As climatic changes proceed leading to increasing environmental variability, empirical studies such as that of Castañeda (2024) are critical in the pursue for a multivariate perspective on trait evolution in scenarios of climate change adaptation. Understanding how tolerance to different environmental stressors may evolve and which factors can act as drivers of that variation will ultimately enable better forecasts of climate change effects on biodiversity in nature. References Castañeda, LE. Cross-tolerance evolution is driven by selection on heat tolerance in Drosophila subobscura. Biorxiv, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology (2024). https://www.doi.org/10.1101/2023.09.05.556367 Bubliy, OA, Loeschcke, V. Correlated responses to selection for stress resistance and longevity in a laboratory population of Drosophila melanogaster. J Evol Biol. 18(4):789-803 (2005). https://www.doi.org/10.1111/j.1420-9101.2005.00928.x. Rodgers, EM, Gomez Isaza, DF. The mechanistic basis and adaptive significance of cross-tolerance: a 'pre-adaptation' to a changing world? J Exp Biol. 226(11):jeb245644 (2023). https://www.doi.org/10.1242/jeb.245644. Schou, MF, Engelbrecht, A, Brand, Z, Svensson, EI, Cloete, S, Cornwallis, CK. Evolutionary trade-offs between heat and cold tolerance limit responses to fluctuating climates. Sci Adv. 8(21):eabn9580 (2022). https://www.doi.org/10.1126/sciadv.abn9580. Singh, K, Arun Samant, M, Prasad, NG. Evolution of cross-tolerance in Drosophila melanogaster as a result of increased resistance to cold stress. Sci Rep. 12(1):19536 (2022). https://www.doi.org/10.1038/s41598-022-23674-z. Stazione, L, Norry, FM, Gomez, FH, Sambucetti, P. Heat knockdown resistance and chill-coma recovery as correlated responses to selection on mating success at high temperature in Drosophila buzzatii. Ecol Evol. 10(4):1998-2006 (2020). https://www.doi.org/10.1002/ece3.6032. | Cross-tolerance evolution is driven by selection of heat tolerance in *Drosophila subobscura* | Luis E. Castañeda | <p>The evolution of heat tolerance is a crucial mechanism for the adaptive response to global warming, but it depends on the genetic variance carried by populations and on the intensity of thermal stress in nature. Experimental selection studies h... | | Adaptation, Experimental Evolution | Pedro Simões | 2023-10-02 14:13:02 |




