Genomic data provides new insights on the demographic history and the extent of recent material transfers in Norway spruce
Jun Chen, Lili Li, Pascal Milesi, Gunnar Jansson, Mats Berlin, Bo Karlsson, Jelena Aleksic, Giovanni G Vendramin, Martin Lascoux
https://doi.org/10.1101/402016
Disentangling the recent and ancient demographic history of European spruce species
Recommended by Jason Holliday based on reviews by 1 anonymous reviewer
Genetic diversity in temperate and boreal forests tree species has been strongly affected by late Pleistocene climate oscillations [2,3,5], but also by anthropogenic forces. Particularly in Europe, where a long history of human intervention has re-distributed species and populations, it can be difficult to know if a given forest arose through natural regeneration and gene flow or through some combination of natural and human-mediated processes. This uncertainty can confound inferences of the causes and consequences of standing genetic variation, which may impact our interpretation of demographic events that shaped species before humans became dominant on the landscape. In their paper entitled "Genomic data provides new insights on the demographic history and the extent of recent material transfers in Norway spruce", Chen et al. [1] used a genome-wide dataset of 400k SNPs to infer the demographic history of Picea abies (Norway spruce), the most widespread and abundant spruce species in Europe, and to understand its evolutionary relationship with two other spruces (Picea obovata [Siberian spruce] and P. omorika [Serbian spruce]). Three major Norway spruce clusters were identified, corresponding to central Europe, Russia and the Baltics, and Scandinavia, which agrees with previous studies. The density of the SNP data in the present paper enabled inference of previously uncharacterized admixture between these groups, which corresponds to the timing of postglacial recolonization following the last glacial maximum (LGM). This suggests that multiple migration routes gave rise to the extant distribution of the species, and may explain why Chen et al.'s estimates of divergence times among these major Norway spruce groups were older (15mya) than those of previous studies (5-6mya) – those previous studies may have unknowingly included admixed material [4]. Treemix analysis also revealed extensive admixture between Norway and Siberian spruce over the last ~100k years, while the geographically-restricted Serbian spruce was both isolated from introgression and had a dramatically smaller effective population size (Ne) than either of the other two species. This small Ne resulted from a bottleneck associated with the onset of the iron age ~3000 years ago, which suggests that anthropogenic depletion of forest resources has severely impacted this species. Finally, ancestry of Norway spruce samples collected in Sweden and Denmark suggest their recent transfer from more southern areas of the species range. This northward movement of genotypes likely occurred because the trees performed well relative to local provenances, which is a common observation when trees from the south are planted in more northern locations (although at the potential cost of frost damage due to inappropriate phenology). While not the reason for the transfer, the incorporation of southern seed sources into the Swedish breeding and reforestation program may lead to more resilient forests under climate change. Taken together, the data and analysis presented in this paper allowed inference of the intra- and interspecific demographic histories of a tree species group at a very high resolution, and suggest caveats regarding sampling and interpretation of data from areas with a long history of occupancy by humans.
References
[1] Chen, J., Milesi, P., Jansson, G., Berlin, M., Karlsson, B., Aleksić, J. M., Vendramin, G. G., Lascoux, M. (2018). Genomic data provides new insights on the demographic history and the extent of recent material transfers in Norway spruce. BioRxiv, 402016. ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/402016
[2] Holliday, J. A., Yuen, M., Ritland, K., & Aitken, S. N. (2010). Postglacial history of a widespread conifer produces inverse clines in selective neutrality tests. Molecular Ecology, 19(18), 3857–3864. doi: 10.1111/j.1365-294X.2010.04767.x
[3] Ingvarsson, P. K. (2008). Multilocus patterns of nucleotide polymorphism and the demographic history of Populus tremula. Genetics, 180, 329-340. doi: 10.1534/genetics.108.090431
[4] Lockwood, J. D., Aleksić, J. M., Zou, J., Wang, J., Liu, J., & Renner, S. S. (2013). A new phylogeny for the genus Picea from plastid, mitochondrial, and nuclear sequences. Molecular Phylogenetics and Evolution, 69(3), 717–727. doi: 10.1016/j.ympev.2013.07.004
[5] Pyhäjärvi, T., Garcia-Gil, M. R., Knürr, T., Mikkonen, M., Wachowiak, W., & Savolainen, O. (2007). Demographic history has influenced nucleotide diversity in European Pinus sylvestris populations. Genetics, 177(3), 1713–1724. doi: 10.1534/genetics.107.077099 "
| Genomic data provides new insights on the demographic history and the extent of recent material transfers in Norway spruce | Jun Chen, Lili Li, Pascal Milesi, Gunnar Jansson, Mats Berlin, Bo Karlsson, Jelena Aleksic, Giovanni G Vendramin, Martin Lascoux | <p>Primeval forests are today exceedingly rare in Europe and transfer of forest reproductive material for afforestation and improvement have been very common, especially over the last two centuries. This can be a serious impediment when inferring ... |  | Evolutionary Applications, Hybridization / Introgression, Population Genetics / Genomics | Jason Holliday | Anonymous, Anonymous | 2018-08-29 08:33:15 | View |
Leaps and bounds: geographical and ecological distance constrained the colonisation of the Afrotemperate by Erica
Michael D Pirie, Martha Kandziora, Nicolai M Nuerk, Nicholas C Le Maitre, Ana Laura Mugrabi de Kuppler, Berit Gehrke, Edward GH Oliver, Dirk U Bellstedt
https://doi.org/10.1101/290791
The colonization history of largely isolated habitats
Recommended by Andrea S. Meseguer based on reviews by Simon Joly, Florian Boucher and 2 anonymous reviewers
The build-up of biodiversity is the result of in situ speciation and immigration, with the interplay between geographical distance and ecological suitability determining the probability of an organism to establish in a new area. The relative contribution of these factors have long interested biogeographers, in particular to explain the distribution of organisms adapted to habitats that remained largely isolated, such as the colonization of oceanic islands or land waters. The focus of this study is the formation of the afrotemperate flora; patches of temperate vegetation separated by thousands of kilometers in Africa, with high levels of endemism described in the Cape region, the Drakensberg range and the high mountains of tropical east Africa [1]. The floristic affinities between these centers of endemism have frequently been explored but the origin of many afrotemperate lineages remains enigmatic [2].
To identify the biogeographic history and drivers of biogeographic movements of the large afrotemperate genus Erica, the study of Pirie and colleagues [3] develops a robust hypothesis-testing approach relying on historical biogeographic models, phylogenetic and species occurrence data. Specifically, the authors test the directionality of migrations through Africa and address the general question on whether geographic proximity or climatic niche similarity constrained the colonization of the Afrotemperate by Erica. They found that the distribution of Erica species in Africa is the result of infrequent colonization events and that both geographic proximity and niche similarity limited geographic movements (with the model that incorporates both factors fitting the data better than null models). Unfortunately, the correlation between geographic and environmental distances found in this study limited the potential evaluation of their roles individually. They also found that species of Erica have dispersed from Europe to African regions, with the Drakensberg Mountains representing a colonization sink, rather than acting as a “stepping stone” between the Cape and Tropical African regions.
Advances in historical biogeography have been recently questioned by the difficulty to compare biogeographic models emphasizing long distance dispersal (DEC+J) versus vicariance (DEC) using statistical methods, such as AIC, as well as by questioning the own performance of DEC+J models [4]. Behind Pirie et al. main conclusions prevails the assumption that patterns of concerted long distance dispersal are more realistic than vicariance scenarios, such that a widespread afrotemperate flora that receded with climatic changes never existed. Pirie et al. do not explicitly test for this scenario based on the idea that these habitats remained largely isolated over time and our current knowledge on African paleoclimates and vegetation, emphasizing the value of arguments based on empirical (biological, geographic) considerations in model comparisons. I, however, appreciate from this study that the results of the biogeographic models emphasizing long distance dispersal, vicariance, and the unconstrained models are congruent with each other and presented together.
Pirie and colleagues [3] bring a nice study on the importance of long distance dispersal and biome shift in structuring the regional floras of Africa. They evidence outstanding examples of radiations in Erica resulting from single dispersal events over long distances and between ecologically dissimilar areas, which highlight the importance of niche evolution and biome shifts in the assembly of diversity. Although we still face important limitations in data availability and model realism, the last decade has witnessed an improvement of our understanding of how historical and environmental triggers are intertwined on shaping biological diversity. I found Pirie et al.’s approach (and analytical framework) very stimulating and hope that will help movement in that direction, providing interesting perspectives for future investigations of other regions.
References
[1] Linder, H.P. 1990. On the relationship between the vegetation and floras of the Afromontane and the Cape regions of Africa. Mitteilungen aus dem Institut für Allgemeine Botanik Hamburg 23b:777–790.
[2] Galley, C., Bytebier, B., Bellstedt, D. U., & Peter Linder, H. (2006). The Cape element in the Afrotemperate flora: from Cape to Cairo?. Proceedings of the Royal Society B: Biological Sciences, 274(1609), 535-543. doi: 10.1098/rspb.2006.0046
[3] Pirie, M. D., Kandziora, M., Nuerk, N. M., Le Maitre, N. C., de Kuppler, A. L. M., Gehrke, B., Oliver, E. G. H., & Bellstedt, D. U. (2018). Leaps and bounds: geographical and ecological distance constrained the colonisation of the Afrotemperate by Erica. bioRxiv, 290791. ver. 5 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/290791
[4] Ree, R. H., & Sanmartín, I. (2018). Conceptual and statistical problems with the DEC+ J model of founder‐event speciation and its comparison with DEC via model selection. Journal of Biogeography, 45(4), 741-749. doi: 10.1111/jbi.13173
| Leaps and bounds: geographical and ecological distance constrained the colonisation of the Afrotemperate by Erica | Michael D Pirie, Martha Kandziora, Nicolai M Nuerk, Nicholas C Le Maitre, Ana Laura Mugrabi de Kuppler, Berit Gehrke, Edward GH Oliver, Dirk U Bellstedt | <p>The coincidence of long distance dispersal and biome shift is assumed to be the result of a multifaceted interplay between geographical distance and ecological suitability of source and sink areas. Here, we test the influence of these factors o... | 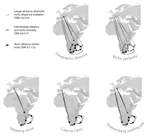 | Phylogeography & Biogeography | Andrea S. Meseguer | | 2018-04-09 10:10:04 | View |
A behavior-manipulating virus relative as a source of adaptive genes for parasitoid wasps
D. Di Giovanni, D. Lepetit, M. Boulesteix, M. Ravallec, J. Varaldi
https://doi.org/10.1101/342758
Genetic intimacy of filamentous viruses and endoparasitoid wasps
Recommended by Ignacio Bravo based on reviews by Alejandro Manzano Marín and 1 anonymous reviewer based on reviews by Alejandro Manzano Marín and 1 anonymous reviewer
Viruses establish intimate relationships with the cells they infect. The virocell is a novel entity, different from the original host cell and beyond the mere combination of viral and cellular genetic material. In these close encounters, viral and cellular genomes often hybridise, combine, recombine, merge and excise. Such chemical promiscuity leaves genomics scars that can be passed on to descent, in the form of deletions or duplications and, importantly, insertions and back and forth exchange of genetic material between viruses and their hosts.
In this preprint [1], Di Giovanni and coworkers report the identification of 13 genes present in the extant genomes of members of the Leptopilina wasp genus, bearing sound signatures of having been horizontally acquired from an ancestral virus. Importantly the authors identify Leptopilina boulardi filamentous virus (LbFV) as an extant relative of the ancestral virus that served as donor for the thirteen horizontally transferred genes. While pinpointing genes with a likely possible viral origin in eukaryotic genomes is only relatively rare, identifying an extant viral lineage related to the ancestral virus that continues to infect an extant relative of the ancestral host is remarkable. But the amazing evolutionary history of the Leptopilina hosts and these filamentous viruses goes beyond this shared genes. These wasps are endoparasitoids of Drosophila larvae, the female wasp laying the eggs inside the larvae and simultaneously injecting venom that hinders the immune response. The composition of the venoms is complex, varies between wasp species and also between individuals within a species, but a central component of all these venoms are spiked structures that vary in morphology, symmetry and size, often referred to as virus-like particles (VLPs).
In this preprint, the authors convincingly show that the expression pattern in the Leptopilina wasps of the thirteen genes identified to have been horizontally acquired from the LbFV ancestor coincides with that of the production of VLPs in the female wasp venom gland. Based on this spatio-temporal match, the authors propose that these VLPs have a viral origin. The data presented in this preprint will undoubtedly stimulate further research on the composition, function, origin, evolution and diversity of these VLP structures, which are highly debated (see for instance [2] and [3]).
References
[1] Di Giovanni, D., Lepetit, D., Boulesteix, M., Ravallec, M., & Varaldi, J. (2018). A behavior-manipulating virus relative as a source of adaptive genes for parasitoid wasps. bioRxiv, 342758, ver. 5 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/342758
[2] Poirié, M., Colinet, D., & Gatti, J. L. (2014). Insights into function and evolution of parasitoid wasp venoms. Current Opinion in Insect Science, 6, 52-60. doi: 10.1016/j.cois.2014.10.004
[3] Heavner, M. E., Ramroop, J., Gueguen, G., Ramrattan, G., Dolios, G., Scarpati, M., ... & Govind, S. (2017). Novel organelles with elements of bacterial and eukaryotic secretion systems weaponize parasites of Drosophila. Current Biology, 27(18), 2869-2877. doi: 10.1016/j.cub.2017.08.019
| A behavior-manipulating virus relative as a source of adaptive genes for parasitoid wasps | D. Di Giovanni, D. Lepetit, M. Boulesteix, M. Ravallec, J. Varaldi | <p>To circumvent host immune response, numerous hymenopteran endo-parasitoid species produce virus-like structures in their reproductive apparatus that are injected into the host together with the eggs. These viral-like structures are absolutely n... |  | Adaptation, Behavior & Social Evolution, Genetic conflicts, Genome Evolution | Ignacio Bravo | | 2018-07-18 15:59:14 | View |
Separate the wheat from the chaff: genomic analysis of local adaptation in the red coral Corallium rubrum
Pratlong M, Haguenauer A, Brener K, Mitta G, Toulza E, Garrabou J, Bensoussan N, Pontarotti P, Aurelle D
https://doi.org/10.1101/306456
Pros and Cons of local adaptation scans
Recommended by Guillaume Achaz based on reviews by Lucas Gonçalves da Silva and 1 anonymous reviewer
The preprint by Pratlong et al. [1] is a well thought quest for genomic regions involved in local adaptation to depth in a species a red coral living the Mediterranean Sea. It first describes a pattern of structuration and then attempts to find candidate genes involved in local adaptation by contrasting deep with shallow populations. Although the pattern of structuration is clear and meaningful, the candidate genomic regions involved in local adaptation remain to be confirmed. Two external reviewers and myself found this preprint particularly interesting regarding the right-mindedness of the authors in front of the difficulties they encounter during their experiments. The discussions on the pros and cons of the approach are very sound and can be easily exported to a large number of studies that hunt for local adaptation. In this sense, the lessons one can learn by reading this well documented manuscript are certainly valuable for a wide range of evolutionary biologists.
More precisely, the authors RAD-sequenced 6 pairs of 'shallow vs deep' samples located in 3 geographical sea areas (Banyuls, Corsica and Marseilles). They were hoping to detect genes involved in the adaptation to depth, if there were any. They start by assessing the patterns of structuration of the 6 samples using PCA and AMOVA [2] and also applied the STRUCTURE [3] assignment software. They show clearly that the samples were mostly differentiated between geographical areas and that only 1 out the 3 areas shows a pattern of isolation by depth (i.e. Marseille). They nevertheless went on and scanned for variants that are highly differentiated in the deep samples when compared to the shallow paired samples in Marseilles, using an Fst outliers approach [4] implemented in the BayeScEnv software [5]. No clear functional signal was in the end detected among the highly differentiated SNPs, leaving a list of candidates begging for complementary data.
The scan for local adaptation using signatures of highly divergent regions is a classical problem of population genetics. It has been applied on many species with various degrees of success. This study is a beautiful example of a well-designed study that did not give full satisfactory answers. Readers will especially appreciate the honesty and the in-depth discussions of the authors while exposing their results and their conclusions step by step.
References
[1] Pratlong, M., Haguenauer, A., Brener, K., Mitta, G., Toulza, E., Garrabou, J., Bensoussan, N., Pontarotti P., & Aurelle, D. (2018). Separate the wheat from the chaff: genomic scan for local adaptation in the red coral Corallium rubrum. bioRxiv, 306456, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/306456
[2] Excoffier, L., Smouse, P. E. & Quattro, J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics, 131(2), 479-491.
[3] Pritchard, J. K., Stephens, M., & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155(2), 945-959.
[4] Lewontin, R. C., & Krakauer, J. (1973). Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics, 74(1), 175-195.
[5] de Villemereuil, P., & Gaggiotti, O. E. (2015). A new FST‐based method to uncover local adaptation using environmental variables. Methods in Ecology and Evolution, 6(11), 1248-1258. doi: 10.1111/2041-210X.12418
| Separate the wheat from the chaff: genomic analysis of local adaptation in the red coral Corallium rubrum | Pratlong M, Haguenauer A, Brener K, Mitta G, Toulza E, Garrabou J, Bensoussan N, Pontarotti P, Aurelle D | <p>Genomic data allow an in-depth and renewed study of local adaptation. The red coral (Corallium rubrum, Cnidaria) is a highly genetically structured species and a promising model for the study of adaptive processes along an environmental gradien... |  | Adaptation, Population Genetics / Genomics | Guillaume Achaz | | 2018-04-24 11:27:40 | View |
Convergent evolution as an indicator for selection during acute HIV-1 infection
Frederic Bertels, Karin J Metzner, Roland R Regoes
https://doi.org/10.1101/168260
Is convergence an evidence for positive selection?
Recommended by Guillaume Achaz based on reviews by Jeffrey Townsend and 1 anonymous reviewer
The preprint by Bertels et al. [1] reports an interesting application of the well-accepted idea that positively selected traits (here variants) can appear several times independently; think about the textbook examples of flight capacity. Hence, the authors assume that reciprocally convergence implies positive selection. The methodology becomes then, in principle, straightforward as one can simply count variants in independent datasets to detect convergent mutations.
In this preprint, the authors have applied this counting strategy on 95 available sequence alignments of the env gene of HIV-1 [2,3] that corresponds to samples taken in different patients during the early phase of infection, at the very beginning of the onset of the immune system. They have compared the number and nature of the convergent mutations to a "neutral" model that assumes (a) a uniform distribution of mutations and (b) a substitution matrix estimated from the data. They show that there is an excess of convergent mutations when compared to the “neutral” expectations, especially for mutations that have arisen in 4+ patients. They also show that the gp41 gene is enriched in these convergent mutations. The authors then discuss in length the potential artifacts that could have given rise to the observed pattern.
I think that this preprint is remarkable in the proposed methodology. Samples are taken in different individuals, whose viral populations were founded by a single particle. Thus, there is no need for phylogenetic reconstruction of ancestral states that is the typical first step of trait convergent analyses. It simply becomes counting variants. This simple counting procedure needs nonetheless to be compared to a “neutral” expectation (a reference model), which includes the mutational process. In this article, the poor predictions of a specifically designed reference model is interpreted as an evidence for positive selection.
Whether the few mutations that are convergent in 4-7 samples out of 95 were selected or not is hard to assess with certainty. The authors have provided good evidence that they are, but only experimental validations will strengthen the claim. Nonetheless, beyond a definitive clue to the implication of selection on these particular mutations, I found the methodological strategy and the discussions on the potential biases highly stimulating. This article is an excellent starting point for further methodological developments that could be then followed by large-scale analyses of convergence in many different organisms and case studies.
References
[1] Bertels, F., Metzner, K. J., & Regoes R. R. (2018). Convergent evolution as an indicator for selection during acute HIV-1 infection. BioRxiv, 168260, ver. 4 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/168260
[2] Keele, B. F., Giorgi, E. E., Salazar-Gonzalez, J. F., Decker, J. M., Pham, K.T., Salazar, M. G., Sun, C., Grayson, T., Wang, S., Li, H. et al. (2008). Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci USA 105: 7552–7557. doi: 10.1073/pnas.0802203105
[3] Li, H., Bar, K. J., Wang, S., Decker, J. M., Chen, Y., Sun, C., Salazar-Gonzalez, J.F., Salazar, M.G., Learn, G.H., Morgan, C. J. et al. (2010). High multiplicity infection by HIV-1 in men who have sex with men. PLoS Pathogens 6:e1000890. doi: 10.1371/journal.ppat.1000890
| Convergent evolution as an indicator for selection during acute HIV-1 infection | Frederic Bertels, Karin J Metzner, Roland R Regoes | <p>Convergent evolution describes the process of different populations acquiring similar phenotypes or genotypes. Complex organisms with large genomes only rarely and only under very strong selection converge to the same genotype. In contrast, ind... |  | Bioinformatics & Computational Biology, Evolutionary Applications, Genome Evolution, Molecular Evolution | Guillaume Achaz | | 2017-07-26 08:39:17 | View |
Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen Islands
Christelle Fraïsse, Anne Haguenauer, Karin Gerard, Alexandra Anh-Thu Weber, Nicolas Bierne, Anne Chenuil
https://doi.org/10.1101/239244
Introgression from related species reveals fine-scale structure in an isolated population of mussels and causes patterns of genetic-environment associations
Recommended by Marianne Elias based on reviews by Thomas Broquet and Tatiana Giraud
Assessing population connectivity is central to understanding population dynamics, and is therefore of great importance in evolutionary biology and conservation biology. In the marine realm, the apparent absence of physical barriers, large population sizes and high dispersal capacities of most organisms often result in no detectable structure, thereby hindering inferences of population connectivity. In a review paper, Gagnaire et al. [1] propose several ideas to improve detection of population connectivity. Notably, using simulations they show that under certain circumstances introgression from one species into another may reveal cryptic population structure within that second species.
The isolated Kerguelen archipelago in the south of Indian Ocean represents a typical situation where the structure of coastal marine organisms is expected to be difficult to detect. In an elegant genomic study, Fraïsse et al. [2] take advantage of introgression from foreign lineages to infer fine-grained population structure in a population of mussels around the Kerguelen archipelago, and investigate its association with environmental variables. Using a large panel of genome-wide markers (GBS) and applying a range of methods that unravel patterns of divergence and gene flow among lineages, they first find that the Kerguelen population is highly admixed, with a major genetic background corresponding to the southern mussel lineage Mytilus platensis introgressed by three northern lineages. By selecting a panel of loci enriched in ancestry-informative SNPs (ie, SNPs highly differentiated among northern lineages) they then detect a fine-scale genetic structure around the Kerguelen archipelago, and identify a major connectivity break. They further show an associating between the genetic structure and environmental variables, particularly the presence of Macrocystis kelp, a marker of habitat exposure to waves (a feature repeatedly evidenced to be important for mussels). While such association pattern could lead to the interpretation that differentiated SNPs correspond to loci directly under selection or linked with such loci, and even be considered as support for adaptive introgression, Fraïsse et al. [2] convincingly show by performing simulations that the genetic-environment association detected can be entirely explained by dispersal barriers associated with environmental variables (habitat-associated connectivity). They also explain why the association is better detected by ancestry-informative SNPs as predicted by Gagnaire et al. [1]. In addition, intrinsic genetic incompatibilities, which reduce gene flow, tend to become trapped at ecotones due to ecological selection, even when loci causing genetic incompatibilities are unlinked with loci involved in adaption to local ecological conditions (Bierne et al. [3]’s coupling hypothesis), leading to correlations between environmental variables and loci not involved in local adaptation. Notably, in Fraïsse et al. [2]’s study, the association between the kelp and ancestry-informative alleles is not consistent throughout the archipelago, casting further doubt on the implication of these alleles in local adaptation.
The study of Fraïsse et al. [2] is therefore an important contribution to evolutionary biology because 1) it provides an empirical demonstration that alleles of foreign origin can be pivotal to detect fine-scale connectivity patterns and 2) it represents a test case of Bierne et al. [3]’s coupling hypothesis, whereby introgressed alleles also enhance patterns of genetic-environment associations. Since genomic scan or GWAS approaches fail to clearly reveal loci involved in local adaptation, how can we disentangle environment-driven selection from intrinsic reproductive barriers and habitat-associated connectivity? A related question is whether we can reliably identify cases of adaptive introgression, which have increasingly been put forward as a mechanism involved in adaptation [4]. Unfortunately, there is no easy answer, and the safest way to go is to rely – where possible – on independent information [5], in particular functional studies of the detected loci, as is for example the case in the mimetic butterfly Heliconius literature (e. g., [6]) where several loci controlling colour pattern variation are well characterized.
References
[1] Gagnaire, P.-A., Broquet, T., Aurelle, D., Viard, F., Souissi, A., Bonhomme, F., Arnaud-Haond, S., & Bierne, N. (2015). Using neutral, selected, and hitchhiker loci to assess connectivity of marine populations in the genomic era. Evolutionary Applications, 8, 769–786. doi: 10.1111/eva.12288
[2] Fraïsse, C., Haguenauer, A., Gerard, K., Weber, A. A.-T., Bierne, N., & Chenuil, A. (2018). Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen Islands. bioRxiv, 239244, ver. 4 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/239244
[3] Bierne, N., Welch, J., Loire, E., Bonhomme, F., & David, P. (2011). The coupling hypothesis: why genome scans may fail to map local adaptation genes. Molecular Ecology, 20, 2044–2072. doi: 10.1111/j.1365-294X.2011.05080.x
[4] Hedrick, P. W. (2013). Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Molecular Ecology, 22, 4606–4618. doi: 10.1111/mec.12415
[5] Ravinet, M., Faria, R., Butlin, R. K., Galindo, J., Bierne, N., Rafajlović, M., Noor, M. A. F., Mehlig, B., & Westram, A. M. (2017). Interpreting the genomic landscape of speciation: a road map for finding barriers to gene flow. Journal of Evolutionary Biology, 30, 1450–1477. doi: 10.1111/jeb.13047.
[6] Jay, P., Whibley, A., Frézal, L., Rodríguez de Cara, M. A., Nowell, R. W., Mallet, J., Dasmahapatra, K. K., & Joron, M. (2018). Supergene evolution triggered by the introgression of a chromosomal inversion. Current Biology, 28, 1839–1845.e3. doi: 10.1016/j.cub.2018.04.072
| Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen Islands | Christelle Fraïsse, Anne Haguenauer, Karin Gerard, Alexandra Anh-Thu Weber, Nicolas Bierne, Anne Chenuil | <p>Reticulated evolution -i.e. secondary introgression / admixture between sister taxa- is increasingly recognized as playing a key role in structuring infra-specific genetic variation and revealing cryptic genetic connectivity patterns. When admi... |  | Hybridization / Introgression, Phylogeography & Biogeography, Population Genetics / Genomics | Marianne Elias | | 2017-12-28 14:16:16 | View |
Field evidence for manipulation of mosquito host selection by the human malaria parasite, Plasmodium falciparum
Amelie Vantaux, Franck Yao, Domonbabele FdS Hien, Edwige Guissou, Bienvenue K Yameogo, Louis-Clement Gouagna, Didier Fontenille, Francois Renaud, Frederic Simard, Carlo Constantini, Frederic Thomas, Karine Mouline, Benjamin Roche, Anna Cohuet, Kounbobr R Dabire, Thierry Lefevre
https://doi.org/10.1101/207183
Malaria host manipulation increases probability of mosquitoes feeding on humans
Recommended by Alison Duncan based on reviews by Olivier Restif, Ricardo S. Ramiro and 1 anonymous reviewer
Parasites can manipulate their host’s behaviour to ensure their own transmission. These manipulated behaviours may be outside the range of ordinary host activities [1], or alter the crucial timing and/or location of a host’s regular activity. Vantaux et al show that the latter is true for the human malaria parasite, Plasmodium falciparum [2]. They demonstrate that three species of Anopheles mosquito were 24% more likely to choose human hosts, rather than other vertebrates, for their blood feed when they harboured transmissible stages (sporozoites) compared to when they were uninfected, or infected with non-transmissible malaria parasites [2]. Host choice is crucial for the malaria parasite Plasmodium falciparum to complete its life-cycle, as their host range is much narrower than the mosquito’s for feeding; P. falciparum can only develop in hominids, or closely related apes [3].
The study only shows this stage-dependent parasite manipulation retrospectively (by identifying host type and parasite stage in mosquitoes after their blood feed [2]). There was no difference in the preferences of infectious (with sporozoites) or un-infectious (infected without sporozoites, or uninfected) mosquitoes between human versus cow hosts in a choice test [2]. This suggests that the final decision about whether to feed occurs when the mosquito is in close range of the host.
This, coupled with previous findings, shows that vector manipulation is a fine-tuned business, that can act at multiple stages of the parasite life-cycle and on many behaviours [4]. Indeed, mosquitoes with non-transmissible Plasmodium stages (oocysts) are more reluctant to feed than sporozoite-infected mosquitoes [5] as vectors can be killed by their host whilst feeding, doing so before they are ready to transmit is risky for the malaria parasite. Thus, it seems that Plasmodium is, to some extent, master of its vector; commanding it not to feed when it cannot be transmitted, to feed when it is ready to be transmitted and to feed on the right type of host. What does this mean for our understanding of malaria transmission and epidemics?
Vantaux et al use a mathematical model, parameterised using data from this experiment, to highlight the consequences of this 24% increase in feeding on humans for P. falciparum transmission. They show that this increase raises the number of infectious bites humans receive from 4 (if sporozoite-infected mosquitoes had the same probability as uninfected mosquitoes) to 14 (an increase in 250%), for mosquitoes with a 15-day life-span, at ratios of 1:1 mosquitoes to humans. Longer mosquito life-spans and higher ratios of mosquitoes to humans further increases the number of infectious bites.
These results [2] have important implications for epidemiological forecasting and disease management. Public health strategies could focus on possible ways to trap sporozoite-infected mosquitoes, mimicking cues they use to locate their human hosts, or identify the behaviour of mosquitoes harbouring non-yet infectious Plasmodium, and trap them before they bite. Moreover, the results of the model show that failing to take into account the preference for humans of sporozoite-infected mosquitoes could underestimate the size of pending epidemics.
An important question previously raised is whether Plasmodium-induced alteration in host behaviour really is manipulation, or just a side-effect of being infected [4,5]. The fact that Vantaux et al show that these altered feeding behaviours increases the likelihood of transmission, in that a sporozoite-infected mosquito is more likely to feed on a human, strongly suggests that it is adaptive for the parasite [2]. Ultimately, to show that it is manipulation would require the identification of molecular factors released by Plasmodium that are responsible for physiological changes in the mosquito [6].
References
[1] Thomas, F., Schmidt-Rhaesa, A., Martin, G., Manu, C., Durand, P., & Renaud, F. (2002). Do hairworms (Nematomorpha) manipulate the water seeking behaviour of their terrestrial hosts? Journal of Evolutionary Biology, 15(3), 356–361. doi: 10.1046/j.1420-9101.2002.00410.x
[2] Vantaux, A., Yao, F., Hien, D. F., Guissou, E., Yameogo, B. K., Gouagna, L.-C., … Lefevre, T. (2018). Field evidence for manipulation of mosquito host selection by the human malaria parasite, Plasmodium falciparum. BioRxiv, 207183 ver 6. doi: 10.1101/207183
[3] Prugnolle, F., Durand, P., Ollomo, B., Duval, L., Ariey, F., Arnathau, C., … Renaud, F. (2011). A Fresh Look at the Origin of Plasmodium falciparum, the Most Malignant Malaria Agent. PLOS Pathogens, 7(2), e1001283. doi: 10.1371/journal.ppat.1001283
[4] Cator, L. J., Lynch, P. A., Read, A. F., & Thomas, M. B. (2012). Do malaria parasites manipulate mosquitoes? Trends in Parasitology, 28(11), 466–470. doi: 10.1016/j.pt.2012.08.004
[5] Cator, L. J., George, J., Blanford, S., Murdock, C. C., Baker, T. C., Read, A. F., & Thomas, M. B. (2013). “Manipulation” without the parasite: altered feeding behaviour of mosquitoes is not dependent on infection with malaria parasites. Proceedings. Biological Sciences, 280(1763), 20130711. doi: 10.1098/rspb.2013.0711
[6] Herbison, R., Lagrue, C., & Poulin, R. (2018). The missing link in parasite manipulation of host behaviour. Parasites & Vectors, 11. doi: 10.1186/s13071-018-2805-9
| Field evidence for manipulation of mosquito host selection by the human malaria parasite, Plasmodium falciparum | Amelie Vantaux, Franck Yao, Domonbabele FdS Hien, Edwige Guissou, Bienvenue K Yameogo, Louis-Clement Gouagna, Didier Fontenille, Francois Renaud, Frederic Simard, Carlo Constantini, Frederic Thomas, Karine Mouline, Benjamin Roche, Anna Cohuet, Kou... | <p>Whether the malaria parasite *Plasmodium falciparum* can manipulate mosquito host choice in ways that enhance parasite transmission toward human is unknown. We assessed the influence of *P. falciparum* on the blood-feeding behaviour of three of... |  | Evolutionary Ecology | Alison Duncan | | 2018-02-28 09:12:14 | View |
Range size dynamics can explain why evolutionarily age and diversification rate correlate with contemporary extinction risk in plants
Andrew J. Tanentzap, Javier Igea, Matthew G. Johnston, Matthew J. Larcombe
https://doi.org/10.1101/152215
Are both very young and the very old plant lineages at heightened risk of extinction?
Recommended by Arne Mooers based on reviews by Dan Greenberg and 1 anonymous reviewer
Human economic activity is responsible for the vast majority of ongoing extinction, but that does not mean lineages are being affected willy-nilly. For amphibians [1] and South African flowering plants [2], young species have a somewhat higher than expected chance of being threatened with extinction. In contrast, older Australian marsupial lineages seem to be more at risk [3]. Both of the former studies suggested that situations leading to peripheral isolation might simultaneously increase ongoing speciation and current threat via small geographic range, while the authors of the latter study suggested that older species might have evolved increasingly narrow niches. Here, Andrew Tanentzap and colleagues [4] dig deeper into the putative links between species age, niche breadth and threat status. Across 500-some plant genera worldwide, they find that, indeed, ""younger"" species (i.e. from younger and faster-diversifying genera) were more likely to be listed as imperiled by the IUCN, consistent with patterns for amphibians and African plants. Given this, results from their finer-level analyses of conifers are initially bemusing: here, ""older"" (i.e., on longer terminal branches) species were at higher risk. This would make conifers more like Australian marsupials, with the rest of the plants being more like amphibians. However, here where the data were more finely grained, the authors detected a second interesting pattern: using an intriguing matched-pair design, they detect a signal of conifer species niches seemingly shrinking as a function of age. The authors interpret this as consistent with increasing specialization, or loss of ancestral warm wet habitat, over paleontological time. It is true that conifers in general are older than plants more generally, with some species on branches that extend back many 10s of millions of years, and so a general loss of suitable habitat makes some sense. If so, both the pattern for all plants (small initial ranges heightening extinction) and the pattern for conifers (eventual increasing specialization or habitat contraction heightening extinction) could occur, each on a different time scale. As a coda, the authors detected no effect of age on threat status in palms; however, this may be both because palms have already lost species to climate-change induced extinction, and because they are thought to speciate more via long-distance dispersal and adaptive divergence then via peripheral isolation.
Given how quickly ranges can change, how hard it is to measure niche breadth, and the qualitatively different time scales governing past diversification and present-day extinction drivers, this is surely not the last word on the subject, even for plants. However, even the hint of a link between drivers of extinction in the Anthropocene and drivers of diversification through the ages is intellectually exciting and, perhaps, even, somehow, of practical importance.
References
[1] Greenberg, D. A., & Mooers, A. Ø. (2017). Linking speciation to extinction: Diversification raises contemporary extinction risk in amphibians. Evolution Letters, 1, 40–48. doi: 10.1002/evl3.4
[2] Davies, T. J., Smith, G. F., Bellstedt, D. U., Boatwright, J. S., Bytebier, B., Cowling, R. M., Forest, F., et al. (2011). Extinction risk and diversification are linked in a plant biodiversity hotspot. PLoS Biology, 9:e1000620. doi: 10.1371/journal.pbio.1000620
[3] Johnson, C. N., Delean S., & Balmford, A. (2002). Phylogeny and the selectivity of extinction in Australian marsupials. Animal Conservation, 5, 135–142. doi: 10.1017/S1367943002002196
[4] Tanentzap, A. J., Igea, J., Johnston, M. G., & Larcombe, M. G. (2018). Range size dynamics can explain why evolutionarily age and diversification rate correlate with contemporary extinction risk in plants. bioRxiv, 152215, ver. 5 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/152215
| Range size dynamics can explain why evolutionarily age and diversification rate correlate with contemporary extinction risk in plants | Andrew J. Tanentzap, Javier Igea, Matthew G. Johnston, Matthew J. Larcombe | <p>Extinction threatens many species, yet few factors predict this risk across the plant Tree of Life (ToL). Taxon age is one factor that may associate with extinction if occupancy of geographic and adaptive zones varies with time, but evidence fo... | 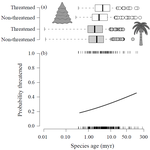 | Macroevolution, Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Arne Mooers | | 2018-02-01 21:01:19 | View |
Parallel pattern of differentiation at a genomic island shared between clinal and mosaic hybrid zones in a complex of cryptic seahorse lineages
Florentine Riquet, Cathy Liautard-Haag, Lucy Woodall, Carmen Bouza, Patrick Louisy, Bojan Hamer, Francisco Otero-Ferrer, Philippe Aublanc, Vickie Béduneau, Olivier Briard, Tahani El Ayari, Sandra Hochscheid, Khalid Belkhir, Sophie Arnaud-Haond, Pierre-Alexandre Gagnaire, Nicolas Bierne
https://doi.org/10.1101/161786
Genomic parallelism in adaptation to orthogonal environments in sea horses
Recommended by Yaniv Brandvain based on reviews by 3 anonymous reviewers
Studies in speciation genomics have revealed that gene flow is quite common, and that despite this, species can maintain their distinct environmental adaptations. Although researchers are still elucidating the genomic mechanisms by which species maintain their adaptations in the face of gene flow, this often appears to involve few diverged genomic regions in otherwise largely undifferentiated genomes. In this preprint [1], Riquet and colleagues investigate the genetic structuring and patterns of parallel evolution in the long-snouted seahorse.
Before investigating specific SNPs plausibly associated with adaptation, the authors first describe genome-wide population structure in the long-snouted seahorse. This species is split into five phenotypically similar, but genetically distinct populations. Two populations reside in the Atlantic Ocean and are geographically structured with one north of the Iberian peninsula and the other around the Iberian peninsula. Two other populations are found in the Mediterranean Sea and are structured by the environment as they correspond to marine and lagoon environments. The genetic clustering of lagoon populations in the Mediterranean, despite the substantial geographic distance between them is quite impressive, and worthy of further study. Finally, a fifth population resides in a lagoon-like habitat in the Black Sea.
The authors then investigate patterns of extreme genomic differentiation among populations, and uncover a remarkable pattern of parallel differentiation in these populations. In an outlier scan, Riquet and colleagues find numerous SNPs in one genomic region that separates northern and southern Atlantic populations. Quiet surprisingly, this same genomic region appears to differentiate populations living in marine and lagoon habitats in the Mediterranean. The idea that parallel patterns of genomic differentiation may underlie adaptation to differing environmental scenarios has not yet received much attention. This paper should change that. This paper is particularity impressive in that the authors uncovered this intriguing pattern with under three hundred SNPs. Future genome scale studies will uncover the genomic basis behind this unusual case of parallelism.
References
[1] Riquet, F., Liautard-Haag, C., Woodall, L., Bouza, C., Louisy, P., Hamer, B., Otero-Ferrer, F., Aublanc, P., Béduneau, V., Briard, O., El Ayari, T., Hochscheid, S. Belkhir, K., Arnaud-Haond, S., Gagnaire, P.-A., Bierne, N. (2018). Parallel pattern of differentiation at a genomic island shared between clinal and mosaic hybrid zones in a complex of cryptic seahorse lineages. bioRxiv, 161786, ver. 4 recommended and peer-reviewed by PCI Evol Biol. doi: 10.1101/161786
| Parallel pattern of differentiation at a genomic island shared between clinal and mosaic hybrid zones in a complex of cryptic seahorse lineages | Florentine Riquet, Cathy Liautard-Haag, Lucy Woodall, Carmen Bouza, Patrick Louisy, Bojan Hamer, Francisco Otero-Ferrer, Philippe Aublanc, Vickie Béduneau, Olivier Briard, Tahani El Ayari, Sandra Hochscheid, Khalid Belkhir, Sophie Arnaud-Haond, Pi... | <p>Diverging semi-isolated lineages either meet in narrow clinal hybrid zones, or have a mosaic distribution associated with environmental variation. Intrinsic reproductive isolation is often emphasized in the former and local adaptation in the la... |  | Hybridization / Introgression, Molecular Evolution, Population Genetics / Genomics, Speciation | Yaniv Brandvain | Sarah Fitzpatrick, Kathleen Lotterhos | 2017-07-11 13:12:40 | View |
Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutations
E. Noël, E. Fruitet, D. Lelaurin, N. Bonel, A. Ségard, V. Sarda, P. Jarne and P. David
https://doi.org/10.1101/273367
Inbreeding compensates for reduced sexual selection in purging deleterious mutations
Recommended by Charles Baer based on reviews by 2 anonymous reviewers
Two evolutionary processes have been shown in theory to enhance the effects of natural selection in purging deleterious mutations from a population (here ""natural"" selection is defined as ""selection other than sexual selection""). First, inbreeding, especially self-fertilization, facilitates the removal of deleterious recessive alleles, the effects of which are largely hidden from selection in heterozygotes when mating is random. Second, sexual selection can facilitate the removal of deleterious alleles of arbitrary dominance, with little or no demographic cost, provided that deleterious effects are greater in males than in females (""genic capture""). Inbreeding (especially selfing) and sexual selection are often negatively correlated in nature. Empirical tests of the role of sexual selection in purging deleterious mutations have been inconsistent, potentially due to the positive relationship between sexual selection and intersexual genetic conflict.
In their preprint, Noël et al. [1] report a cleverly designed, and impressively long-term, experimental evolution study designed to tease apart the relative contributions of selfing and sexual selection in purging deleterious mutations, using the self-compatible hermaphroditic snail Physa acuta. Hermaphroditism relieves at least some of the potential conflict between males and females because each individual expresses traits of each sex. The authors report a 50-generation (ten years!) evolution experiment with four experimental treatments: Control (C), in which snails reproduced by mass mating (allowing sexual selection) and the next generation was sampled randomly from offspring in proportion to maternal family size; Male-selection (M) in which snails reproduced by mass mating but maternal family size was held constant, removing the opportunity for fertility selection; Female fertility selection (F) in which snails mated monogamously but fertility selection was imposed, and selfing (S), in which snails reproduced by selfing every other generation, alternating with monogamy + fertility selection. Juvenile survival was taken as the proxy for fitness and was measured for offspring of self-fertilization and of outcross matings. Each line type (C, M, F, S) was replicated twice.
The results are enviably clear-cut: after 50 generations of evolution, outcross fitness dropped precipitously in the F treatment (monogamy+female fertility selection) and remained at ancestral levels in the other three treatments. Clearly, sexual selection in males is more efficient at purging deleterious alleles than is female fertility selection. Similarly, inbreeding depression was reduced in the S lines relative to the other treatments, indicating that, unsurprisingly, deleterious recessive mutations of large effect are purged under strong inbreeding. Outcross fitness in the S lines did not decline, in contrast to the F lines, which indicates that deleterious mutations are on average slightly recessive.
Taken as a whole, this study by Noël et al. [1] provides a compelling empirical demonstration of the efficacy of both sexual selection and strong inbreeding as mechanisms of purging, and implicates sexual conflict as a potentially important factor in studies in which relaxation of sexual selection fails to result in purging.
References
[1] Noël, E., Fruitet, E., Lelaurin, D., Bonel, N., Segard, A., Sarda, V., Jarne, P., & David P. (2018). Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutations. bioRxiv, 273367, ver. 3 recommended and peer-reviewed by PCI Evol Biol. doi: 10.1101/273367
| Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutations | E. Noël, E. Fruitet, D. Lelaurin, N. Bonel, A. Ségard, V. Sarda, P. Jarne and P. David | <p>This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (https://dx.doi.org/10.24072/pci.evolbiol.100055). Theory and empirical data showed that two processes can boost selection against deleterious mutations, ... |  | Adaptation, Experimental Evolution, Reproduction and Sex, Sexual Selection | Charles Baer | Anonymous | 2018-03-01 08:12:37 | View |





